Fe3O4@SiO2@Ag纳米复合材料的制备及其对苯唑西林的表面增强拉曼光谱检测
2020-03-20徐娅娟李利军黄文艺
徐娅娟 黄 烁 李利军 程 昊,黄文艺 冯 军*,3
(1.广西科技大学医学院广西柳州 545000;2.广西糖资源绿色加工重点实验室广西科技大学生物与化学工程学院广西柳州 545006;3.蔗糖产业省部共建协同创新中心广西南宁 530004)
1977年,Jeanmaire和Van Duyne在粗糙的Ag电极表面发现拉曼信号强度异常增强,可以达到百万倍[1]。进一步研究发现,少量贵金属元素如Au、Ag和Cu自由电子类金属材料表面,可以表现出明显的拉曼增强效应,即表面增强拉曼光谱(SERS)[2]。由于Ag纳米粒子(AgNPs)容易制备,且与Au、Cu等其它材料相比,可获得高出近100倍的增强效果[3],因而成为SERS中最常见的研究对象。Fe3O4磁性纳米粒子(MNPs)具有超顺磁性,在复杂基体中具有很好的分离富集等功能,已广泛应用于生物医学[4]、成像[5]、生物分离[6]及催化[7]等领域。但是裸Fe3O4纳米粒子易于氧化或聚集,且可溶于酸性溶液中,其活性基团不足[8],限制了它的应用。有文献报道[9,10],Fe3O4@SiO2纳米复合物可以屏蔽磁偶极相互作用,避免颗粒团聚,提高其稳定性,已被广泛用于SERS活性基底对目标物进行快速检测。但这种检测方式繁琐,检测重复性差。同时,AgNPs相互之间所产生的“热点”少,SERS活性较低[11,12],灵敏度不高。因此,Hu等人制备出了Fe3O4@SiO2@AgNPs复合物作为SERS基底,并对其性能进行研究,但该方法制备过程繁琐、耗时,而且检测效果不甚理想[13]。因此,改进检测重现性、提高灵敏度对于SERS的快速检测仍然是亟待解决的问题。
本研究采用层层自组装方法,制备出一种高性能Fe3O4@SiO2@Ag纳米复合材料,该材料以Fe3O4为核,以SiO2为壳层,构建Fe3O4@SiO2“核-壳”结构,利用Ag种的各向同性生长和聚乙烯吡咯烷酮(PVP)的稳定作用形成AgNPs沉积在核壳结构上,得到分散性良好、磁响应好、灵敏度高、集成化Fe3O4@SiO2@Ag核壳结构,并通过扫描电镜、透射电镜、能谱及红外光谱等手段对其进行了性能表征。Fe3O4@SiO2@Ag作为SERS活性基底时,具有良好的超顺磁性能,稳定性好,AgNPs在核壳结构上的均匀沉积,提高了检测重复性。在此基础上,本文建立了苯唑西林的SERS快速检测方法。该方法简单、快速、高度集成、重现性好、灵敏度高,已经成功用于各种剂型药物中苯唑西林的快速测定。
1 实验部分
1.1 仪器与试剂
Hitachi S-4800冷场发射扫描电子显微镜(日本,日立公司);JEM 2100透射电子显微镜(日本,电子株式会社);50max能谱仪(英国,牛津仪器);Bruker D8A A25 X射线衍射仪(德国,布鲁克公司);傅里叶变换红外光谱仪(美国,Perkin Elmer公司);XploRAPLUS激光共聚焦拉曼光谱仪(日本,Horiba公司);XS105DU电子天平(梅特勒-托利多国际贸易(上海)有限公司)。
苯唑西林标准品(20 mg/branch,上海邦景实业有限公司)溶液:精确称取4 mg苯唑西林标准品,用HCl(pH=3.0)定容于10 mL容量瓶中,并将上述溶液稀释至所需浓度(10-3,10-4,10-5,10-6,10-7,10-8,10-9,10-10,10-11mol/L)待用。FeCl3·6H2O(天津科密欧化学试剂有限公司),AgNO3(国药集团化学试剂有限公司),乙二醇(广东光华科技股份有限公司),硅酸四乙酯(阿拉丁公司),聚乙二醇4000(西陇化工股份有限公司),NaAc(西陇科学股份有限公司),氨水(西陇科学股份有限公司),乙醇(成都市科隆化学品有限公司),聚乙烯吡咯烷酮(PVP,国药集团化学试剂有限公司),所用试剂均为分析纯或优级纯。实验用水为实验室自制纯化水。
苯唑西林片(0.25 g/tablet,四川制药制剂有限公司),苯唑西林胶囊(0.25 g/pellet,四川制药制剂有限公司),苯唑西林粉针剂(0.5 g/branch,瑞阳制药有限公司)。
1.2 材料制备
1.2.1 Fe3O4的制备采用Modi化溶剂热反应[14]合成Fe3O4磁性纳米粒子。将1.35 g FeCl3·6H2O溶于40 mL乙二醇中,磁力搅拌30 min。然后在所得溶液中加入2.7 g NaAc和1.0 g PEG 4000,搅拌至反应物完全溶解,之后将该溶液转移至高压反应釜中,放入烘箱中于210 ℃反应6 h,取出后冷却至室温。制备的Fe3O4纳米粒子用磁铁收集,用水和乙醇各洗涤3次,放入60 ℃真空干燥箱干燥6 h,待用。
1.2.2 Fe3O4@SiO2纳米粒子的制备称取0.5 g上述制备的Fe3O4,分散到60 mL无水乙醇以及12 mL水的混合溶液中,超声20 min后,滴加2.8 mL氨水。然后在机械搅拌下逐滴加入2 mL TEOS(控制滴加速度为100 μL/min)。脉冲超声反应240 min,用磁铁收集所得粒子,分别用乙醇和水各洗涤3次。放入60 ℃真空干燥箱中干燥12 h后,待用。
1.2.3 Fe3O4@SiO2@Ag纳米复合材料的制备[15]将0.2 g AgNO3溶解在4 mL水中,加入400 μL氨水,超声10 min。将30 mg Fe3O4@SiO2分散于该溶液中,然后加入溶解于无水乙醇(26 mL)的PVP(0.5 g)溶液,超声20 min,将反应物转移至高压反应釜中,于烘箱中120 ℃反应4 h。反应结束后,在磁石吸附下用水和无水乙醇交替洗涤各3次后,置于60 ℃真空干燥箱中干燥5 h,待用。
1.3 样品制备及拉曼光谱采集参数
吸取待测样品溶液500 μL,加入10 mg制备的纳米复合材料,超声1 h后,将混合溶液滴在外加磁场的石英玻片上晾干,每个样品采集3次拉曼光谱。光谱参数:波长为638 nm,积分时间为5 s,平均次数为1次。
2 结果与讨论
2.1 扫描电镜(SEM)、透射电镜(TEM)及能谱(EDS)表征
图1(a~f)分别为Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@Ag的SEM、TEM和EDS表征结果。由图1a可知,Fe3O4(200 nm)大小均一、分散性好、表面光滑,且在能谱(图1b)中只出现Fe、O元素的谱峰,说明Fe3O4表面并没有其他杂质。用SiO2对Fe3O4进行包覆后,Fe3O4形成明显的“核-壳”结构,外层为透明的硅壳层(20 nm),内层为较暗的Fe3O4核(图1c右上),且在能谱(图1d)中出现了Si元素的谱峰,这表明SiO2已成功包覆Fe3O4纳米粒子。由Fe3O4@SiO2@Ag的SEM(图1e)和TEM(图1e右上)图可以看到,AgNPs 均匀的点缀在Fe3O4@SiO2表面,很少观察到较大的Ag颗粒,形成了具有纳米级粗糙度的连续壳层,且在能谱(图1f)中出现了Ag元素的谱峰,进一步证明Fe3O4@SiO2@Ag得以成功制备。
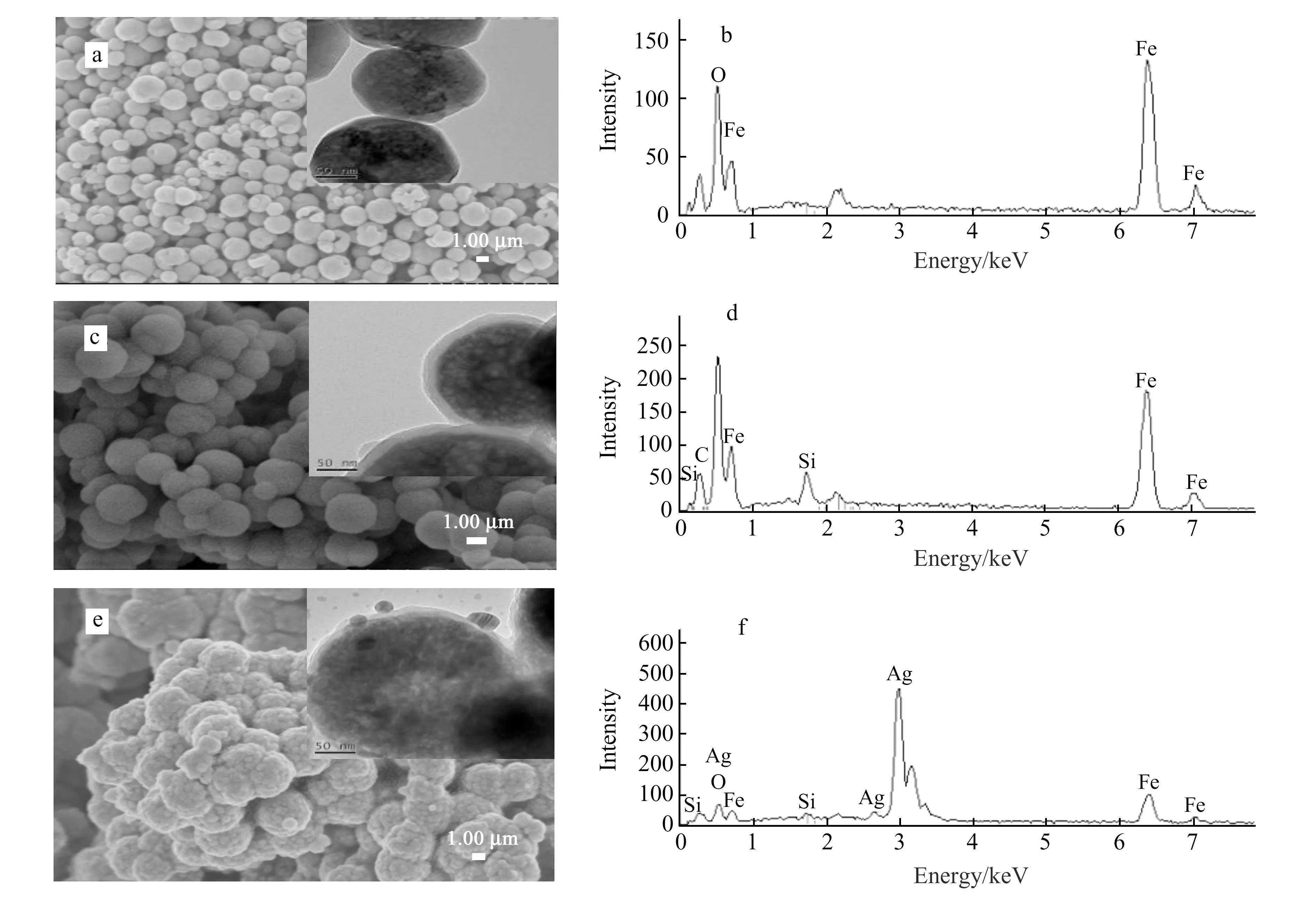
图1 扫描电镜(SEM)、透射电镜(TEM)以及能谱(EDS)图:(a、b)Fe3O4;(c、d)Fe3O4@SiO2;(e、f)Fe3O4@SiO2@AgFig.1 SEM,TEM and EDS results:(a,b)Fe3O4;(c,d)Fe3O4@SiO2;(e,f)Fe3O4@SiO2@Ag
2.2 X射线衍射(XRD)表征
采用XRD分别对Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@Ag进行了表征。如图2所示,Fe3O4磁性纳米粒子(2a)和Fe3O4@SiO2(2b)均在2θ为18.4°、30.1°、35.5°、37.2°、43.1°、53.6°、57.0°、62.6°、74.1°处出现衍射峰。与标准PDF卡片(PDF01-1111)对比可知,这些峰分别归属于Fe3O4立方体结构的(111)、(220)、(311)、(222)、(400)、(422)、(211)、(440)和(533)晶面[16]。这说明在SiO2包覆Fe3O4的过程中,并没有破坏Fe3O4磁性纳米粒子的晶型结构。Fe3O4@SiO2@Ag(图3c)与Fe3O4的XRD谱图对比,前者分别在2θ为38.2°、44.3°、64.5°和77.4°处出现新的衍射峰。与标准卡片(COD 9011607)对比后发现,这些衍射峰分别对应Ag的(111)、(200)、(220)和(311)晶面,并且没有破坏核壳结构内部Fe3O4的晶型结构。这与前述SEM、TEM、EDS表征结果相符,表明Fe3O4@SiO2@Ag成功制备。
2.3 傅里叶变换红外(FT-IR)光谱表征
图3分别为Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@Ag的红外(IR)光谱图。对比图3a、3c可以发现,Fe3O4表面包覆SiO2后,在1 056 cm-1处出现明显的特征峰,该吸收峰是由Si-O键伸缩振动引起的[17]。进一步沉积Ag后(图3b)发现Si-O键吸收峰强度有所减弱,这是因为Fe3O4@SiO2表面沉积Ag较厚且覆盖均匀引起的。这与Fe3O4@SiO2@Ag的SEM和TEM所观察到的结果一致。
2.4 Fe3O4@SiO2@Ag SERS基底对药物中苯唑西林的检测
图4为不同基底的拉曼光谱图。从图4a、4b、4c可以看出Fe3O4和Fe3O4@SiO2、Fe3O4@SiO2@Ag 3种空白基底并没有SERS信号,说明对苯唑西林的SERS信号没有干扰。由图4d可见,苯唑西林在683 cm-1、1 036 cm-1、1 196 cm-1、1 501 cm-1、1 636 cm-1处均出现了拉曼谱峰,而在图4f中在1 028 cm-1(羧基C-O键伸缩振动引起)[18]、1 621 cm-1(苯环C-C键伸缩键振动引起)[24]处拉曼谱峰显著增强,与标准品(图4d)比较后发现,这两处峰发生了轻微红移(大约10 cm-1)。这可能是由于基底的影响导致的,但并不影响该Fe3O4@SiO2@Ag基底对苯唑西林的检测。
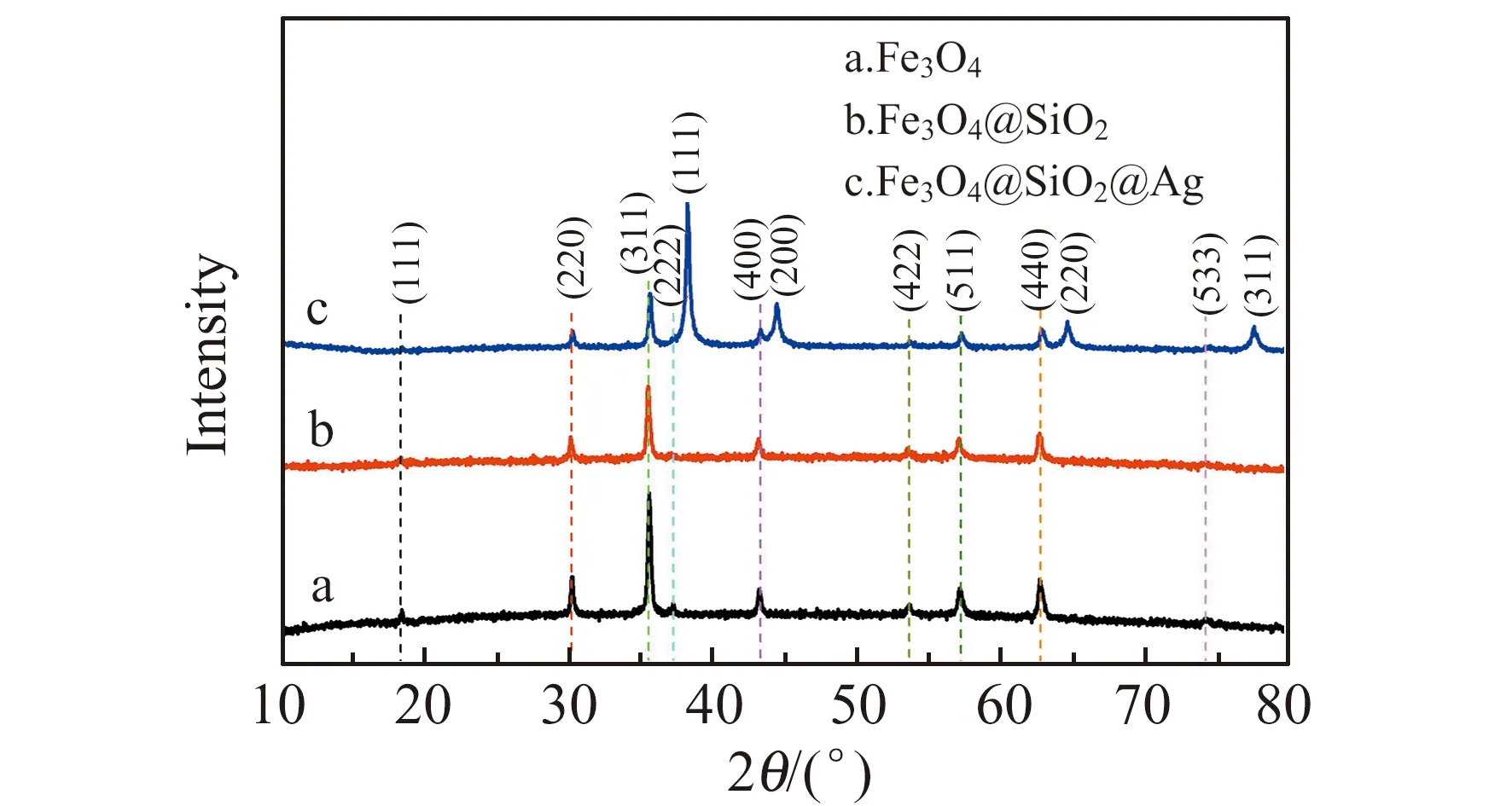
图2 Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@Ag的X射线衍射(XRD)谱图Fig.2 XRD patterns of Fe3O4,Fe3O4@SiO2 and Fe3O4-@SiO2@Ag
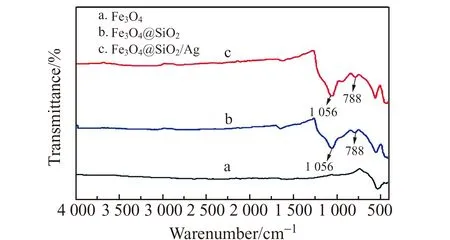
图3 Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@Ag的红外(IR)光谱图Fig.3 IR spectra of Fe3O4,Fe3O4@SiO2 and Fe3O4-@SiO2@Ag
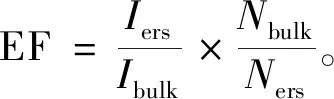
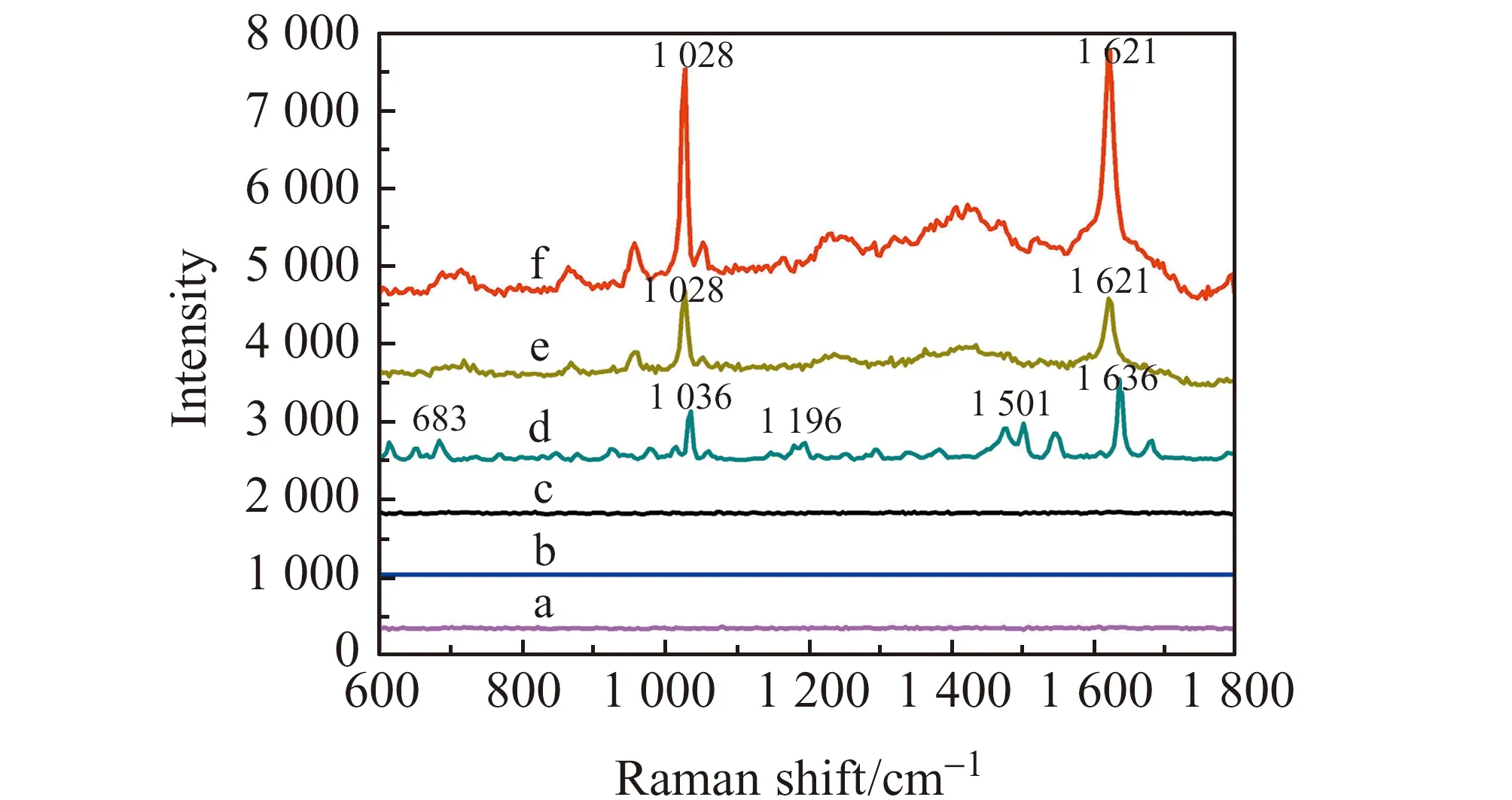
图4 Fe3O4(a)、Fe3O4@SiO2(b)、Fe3O4@SiO2@Ag(c)、苯唑西林标准品(d)、Ag-苯唑西林(10-3 mol/L)(e)和Fe3O4@SiO2@Ag-苯唑西林(10-3 mol/L)(f) SERS图Fig.4 SERS of Fe3O4(a),Fe3O4@SiO2(b),Fe3O4@SiO2@Ag(c),Oxacillin standard;(d)Ag-oxacillin(10-3 mol/L)(e) and Fe3O4@SiO2@Ag-Oxacillin(10-3 mol/L)(f)
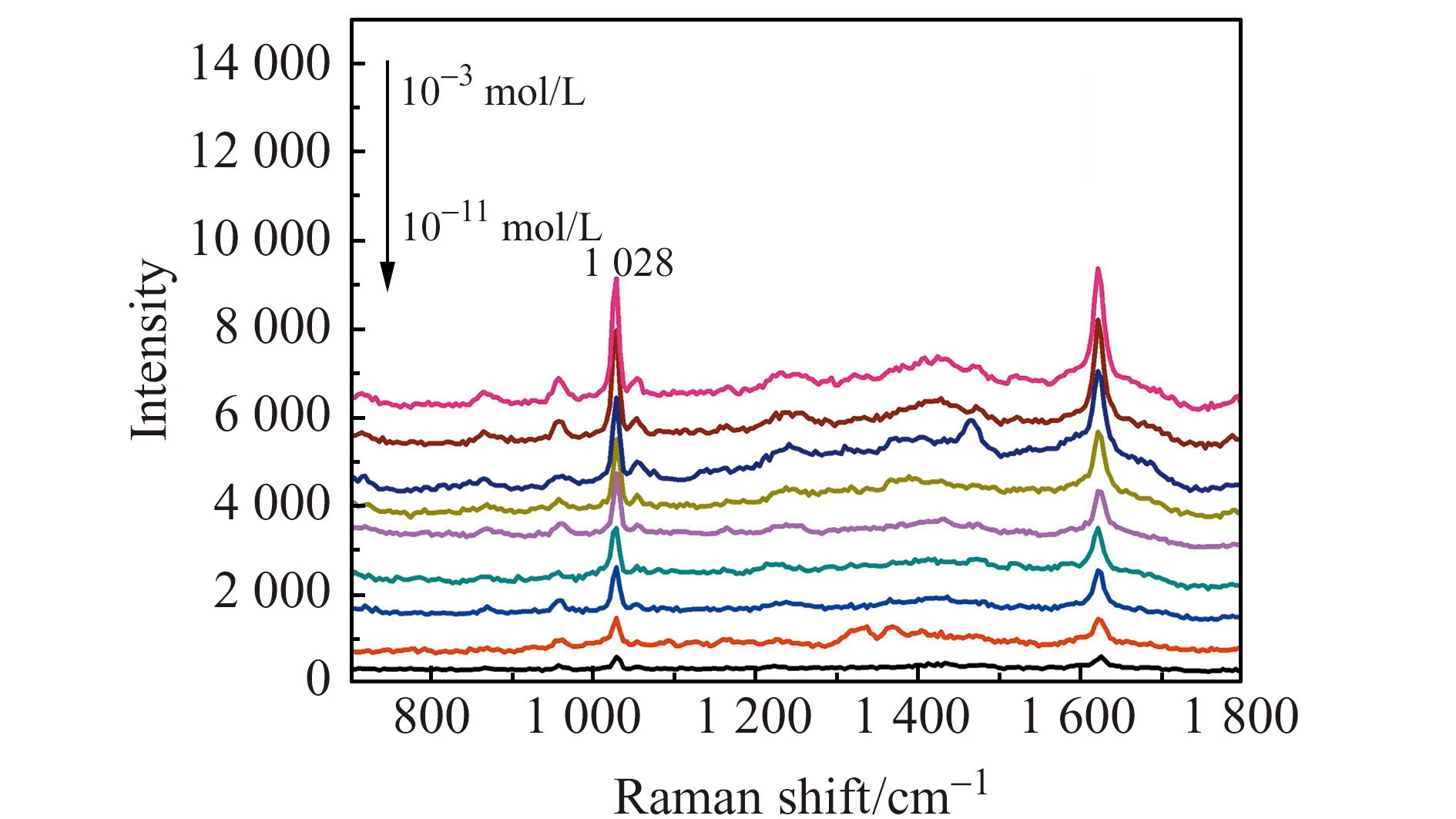
图5 不同浓度的苯唑西林在Fe3O4@SiO2@Ag基底上的SERS图Fig.5 SERS of different concentrations of oxacillin on Fe3O4@SiO2@Ag substrates
本文以1 028 cm-1处的拉曼峰强度作为I值,计算Fe3O4@SiO2@Ag基底对苯唑西林的增强因子为2.5×105。
光谱重现性是影响该方法有效性和实用性的重要因素[21]。对6份平行配制的1.0×10-9mol/L苯唑西林溶液进行了拉曼检测(图6A),考察苯唑西林在该基底上的光谱重现性,发现其在1 028 cm-1处拉曼信号强度的相对标准偏差(RSD)为7.2%(图6B),说明该方法重现性良好。
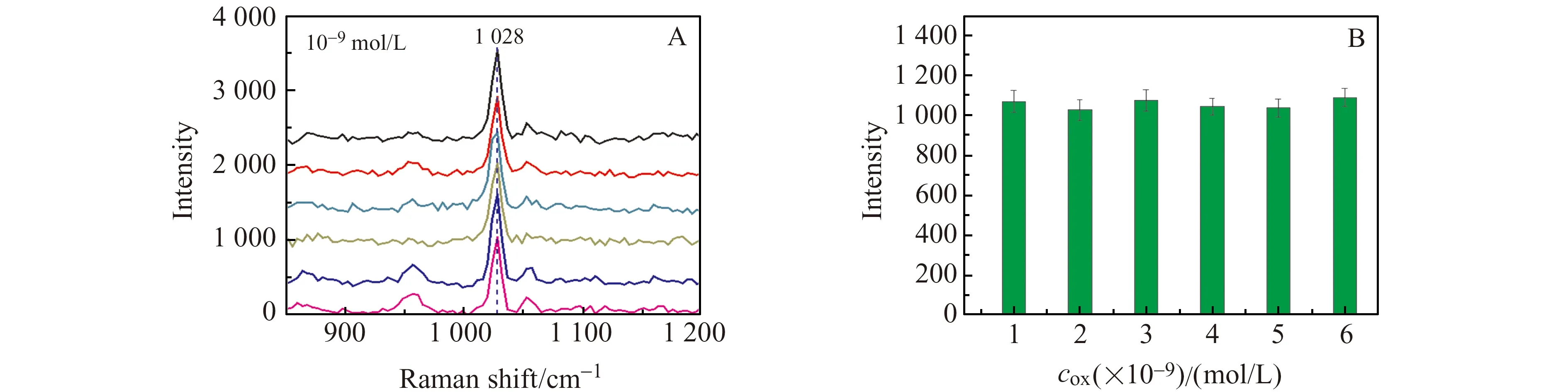
图6 (A)6组苯唑西林(10-9 mol/L)的SERS图;(B)6份苯唑西林(10-9 mol/L)溶液在1 028 cm-1处的拉曼峰(误差条表示三次测量值的标准差)Fig.6 (A) SERS of six groups of oxacillin(10-9 mol/L);(B) Raman peaks of six groups of oxacillin(10-9 mol/L) at 1 028 cm-1(error bars indicate the standard deviation of the three measurements)
2.5 不同剂型苯唑西林的SERS检测
本文对苯唑西林粉针剂、胶囊、片剂分别进行了检测。首先,将苯唑西林胶囊(去其外壳)、片剂进行研磨;然后,分别称取4 mg 3种不同剂型的苯唑西林样品,用HCl(pH=3.0)定容于10 mL容量瓶中,待用。图7为不同剂型的苯唑西林在Fe3O4@SiO2@Ag基底上的SERS图。从图中可以看出,不同剂型的苯唑西林与标准品对比,在1 028 cm-1、1 621 cm-1处均有较好的拉曼吸收,且胶囊的拉曼吸收相对较强,其次是片剂,粉针剂最弱。这可能是由于胶囊中苯唑西林含量较高,而片剂与粉针剂中含量较少。有研究表明,带有巯基、氨基、苯环结构的分子与Ag表面具有很强的化学亲和力[22]。我们所制备的基底材料之所以能够展现出优异的抗干扰能力,归因于该基底与目标物的氨基、苯环结合之后,利用Fe3O4的超顺磁性,可从复杂样品溶液中快速分离富集苯唑西林分子,从而排除制剂中辅料的干扰。
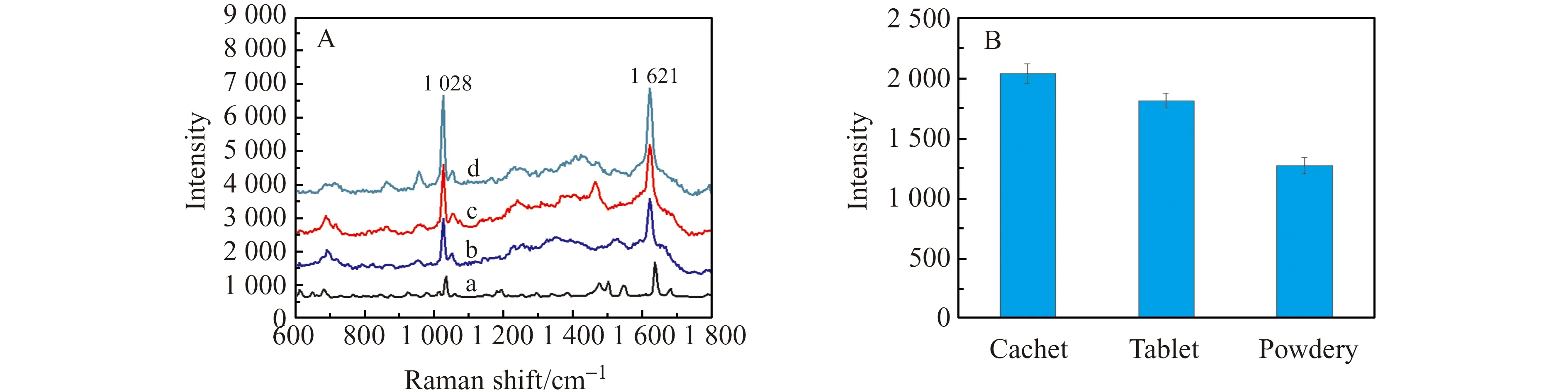
图7 (A)苯唑西林标准品(a)、胶囊(b)、片剂(c)和粉针剂(d)的SERS图;(B)拉曼峰位于1 028 cm-1处的误差分析图Fig.7 (A)SERS of oxacillin standard (a),cachet(b),tablet(c) and powdery(d);(B)Error analysis diagram of Raman peak at 1 028 cm-1
3 结论
本文采用层层自组装方法,制备了超顺磁性、高SERS活性的Fe3O4@SiO2@Ag基底,并对其形貌与结构进行了表征。该材料分散良好、磁响应好、灵敏度高、集成化,可对待测目标物进行有效分离及富集,显著提高了检测灵敏度及抗干扰能力。在该SERS活性基底之上,苯唑西林检测限达1.0×10-11mol/L,显示该基底在食品、药品的SERS快速检测方面具有一定应用价值。
