通过式净化-高效液相色谱-串联质谱法测定动物源性食品中42种兽药残留
2020-03-21张林田陆奕娜黄学泓李冠斯吕坚铃
张林田* 陆奕娜 黄学泓 林 文 李冠斯 吕坚铃
(汕头海关检验检疫技术中心广东汕头 515041)
兽药残留是指动物在使用了兽药后,其蓄积或储存在动物细胞、组织或器官内的药物原型、有毒性的代谢物或杂质,所以监控不同种类的兽药在动物源性产品中的残留是确保食品安全的有效手段。但是目前兽药残留分析的标准方法一般只能分析化学结构类似的同族药物[1-4],因此,建立多种类、多兽药残留的分析方法具有很强的实用意义。
磺胺类(SAs)药物是具有对氨基苯磺酰胺结构的一类抗生素的总称,用于预防和治疗细菌感染性疾病,因具有疗效好、抗菌谱广、性质稳定、价格低廉等优点而被广泛应用于畜牧和水产养殖业。磺胺增效剂是指含有5-取代苄基-2,4-二氨基嘧啶的一类化合物,为广谱抑制剂,多与磺胺合用,使细菌的叶酸代谢受到双重阻断,抗菌作用可增强数10倍[5]。喹诺酮类(QNs)药物是人工合成的含有吡酮酸(1,4-二氢-4-氧吡啶-3-羧酸)基本结构的一类药物,具有抗菌谱广、高效、低毒、组织穿透力强、价格低廉等特点,是兽医临诊和水产养殖中最重要的抗感染药物之一,被大量用于预防和治疗动物疾病及促生长。兽药的过量使用可导致细菌产生抗药性,其残留会通过食物链对人体产生直接毒副作用。世界各国都规定了兽药在动物源性食品中的最大残留限量,我国规定动物源性食品中SAs的最高残留限量为100 μg/kg,磺胺增效剂(三甲氧苄氨嘧啶)为50 μg/kg,猪肌肉中恩诺沙星为100 μg/kg[6]。本文建立了通过式净化-高效液相色谱-串联质谱法测定动物源性食品中SAs、磺胺增效剂及QNs共42种兽药残留,方法具有简单、快捷、灵敏度高、定性准确等特点。
1 实验部分
1.1 仪器与试剂
API4000 plus型三重四极杆质谱仪(美国,AB公司);30AD系列高效液相色谱仪(日本,岛津公司);IKA T25数显型高速分散机(德国,IKA公司);Eppendorf 5810R冷冻高速离心机(德国,Eppendorf公司);MGS-2200型氮吹浓缩仪(日本,东京理化公司)。
45种药物标准品的纯度在93.0%~99.0%范围内,均购于德国Dr.Ehrenstorfer公司,使用时按实际成分进行折算。42种兽药及3种内标(具体名称见表1)标准储备溶液:取适量标准品,用甲醇溶解,分别配制成质量浓度为0.5 g/L的标准储备溶液,置棕色容量瓶中,于-18 ℃避光保存。使用时用甲醇稀释成浓度为1.0 mg/L的混合标准使用液。甲醇、乙腈(色谱纯,美国TEDIA公司);甲酸(色谱纯,美国TEDIA公司);提取液(0.1%甲酸乙腈):吸取1 mL HAc,转移入1 000 mL容量瓶,用乙腈定容并摇匀;0.1%甲酸溶液:准确吸取1 mL甲酸,转移入1 000 mL容量瓶,用水定容并摇匀,作为本实验的流动相;溶解液:乙腈-0.1%甲酸水溶液(20∶80,V/V)。实验用水为(GB/T6682)规定的一级水。
1.2 样品前处理
1.2.1 样品提取准确称取5 g样品(精确至0.01 g)于50 mL聚丙烯离心管中,加入约5 g无水Na2SO4、12 mL乙腈-1%甲酸溶液、100 μL 1.0 mg/L同位素内标混合液,12 000 r/min均质提取30 s后,振荡15 min,4 000 r/min离心5 min,收集上清液。取12 mL 0.1%甲酸乙腈洗涤刀头10 s,洗涤液倒入残渣,用玻棒搅散,旋涡1 min,振荡提取15 min,4 000 r/min离心5 min,合并上清液,用提取液定容至25.0 mL。
1.2.2 提取液净化PRiME HLB固相萃取柱置于固相萃取装置上,取样品提取液5 mL过柱,自然滴出,滴干后用0.8 mL乙腈-甲醇(9+1)淋洗小柱两次,收集全部流出液于10 mL试管中,40 ℃氮气吹干,加入1.0 mL样品溶解液,旋涡混合3 min,转移至1.5 mL离心管中,4 ℃、12 000 r/min 离心5 min,取下层清液过0.22 μm滤膜,供高效液相色谱-串联质谱分析。
1.2.3 空白基质标准溶液的制备称取7份阴性样品5 g,分别加入0.5 mg/L混合标准溶液0、10、50、100、200、500、1 000 μL及100 μL 1.0 mg/L同位素内标溶液,以下按“1.2.1”及“1.2.2”操作步骤进行。
1.3 色谱-质谱条件
1.3.1 色谱条件Waters Atlantis T3色谱柱(150 mm×4.6 mm,3 μm;美国Waters公司),流动相A为乙腈,B为0.1%甲酸溶液。流动相梯度洗脱程序为:起始20%A;0~15 min,线性升至30%A;15~21 min,线性升至70%A,保持4 min;0.2 min降至20%A,保持3.8 min。柱温30 ℃;流速400 μL/min;进样体积10 μL。
1.3.2 质谱条件电喷雾电离正离子(ESI+)模式;动态多反应监测(dMRM)模式检测;雾化气压力(GSI):60 psi;辅助气压力:50 psi;气帘气压力(CUR):25 psi;离子源温度:600 ℃;电喷雾电压(IS):5 500 V;MS1及MS2均为Unit单位分辨率;碰撞气压力(CAD):6 psi;MRM检测窗口:120 s;化合物扫描时间:2 s。42种药物及3种同位素内标的dMRM质谱相关参数见表1。
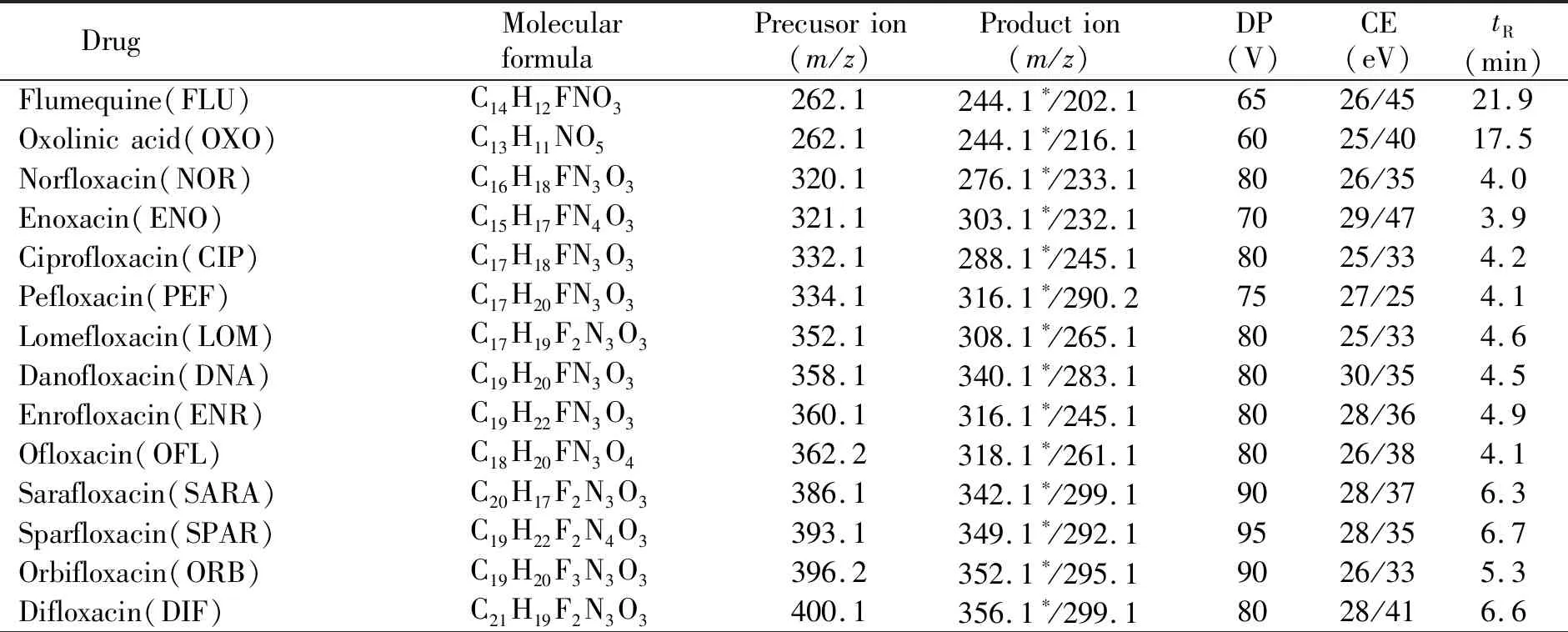
表1 42种兽药的动态多反应监测(dMRM)离子对及质谱参数Table 1 Dynamic multiple reaction monitoring transition and mass spectrometry parameters of representative compounds within 42 veterinary drugs
(续表1)

Drug Molecular formulaPrecusorion(m/z)Production(m/z)DP(V)CE(eV)tR(min)Fleroxacin(FLE)C17H18F3N3O3370.1326.1∗/269.18027/364.1Nalidixicacid(NAL)C12H12N2O3233.0215.0∗/187.02119/3521.4Marbofloxacin(MAR)C17H19FN4O4363.172.0∗/320.18046/233.9Enrofloxacin-D5(ENR-D5)C19H18D5CLFN3O3365.3321.3100324.9Sulfanilamide(SA)C6H8N2O2S173.0156.0∗/108.03010/214.2Sulfaguanidine(SG)C7H10N4O2S215.0156.0∗/92.05215/313.7Sulfadiazine(SD)C10H10N4O2S251.1156.0∗/92.16322/386.1Sulfapyridine(SPD)C11H11N3O2S250.1156.0∗/108.06523/346.7Sulfathiazole(STZ)C9H9N3O2S2256.1156.1∗/92.16022/376.1Diaveridine(DVD)C13H16N4O2261.2245.3∗/123.16535/333.7Sulfamerazine(SMR)C11H12N4O2S265.1156.1∗/172.07324/247.7Sulfamethazine(SMTZ)C11H12N4O3S279.1186.0∗/124.17525/353.9Trimethoprlm(TMP)C14H18N4O3291.1230.1∗/123.19533/343.8Sulfacetamide(SAM)C8H10N2O3S215.0156.0∗/92.05215/315.7Sulfadiazine-13C6(SDZ-13C6)#C413C6H10N4O2S257.1162.160236.1Sulfameter(SMT)C11H12N4O3S281.0156.0∗/126.17525/259.1Sulfadimidine(SDM)C12H14N4O2S279.1124.1∗/1867527/359.1Sulfamonomethoxine(SMM)C11H12N4O3S281.0156.0∗/126.17525/2711.3Sulfamethoxypyridazine(SMPD)C11H12N4O3S281.0156.0∗/126.17525/279.5Sulfamethoxazole(SMZ)C10H11N3O3S254.1156.0∗/108.07023/3214.4Sulfamoxol(SMXL)C11H13N3O3S268.1156.0∗/113.17021/237.8Sulfabenzamide(SBZ)C13H12N2O3S277.1156.0∗/108.16019/3218.8Sulfachloropyridazine(SCP)C10H9CLN4O2S285.1156.0∗/92.16532/2212.6Sulfaquinoxaline(SQX)C14H12N4O2S301.1156.1∗/92.18024/4119.6Sulfadoxine(SDX)C12H14N4O4S311.0156.1∗/108.18031/3814Sulfaphenazole(SPA)C15H14N4O2S315.0156.0∗/222.18429/2819.9Sulfisoxazole(SFZ)C11H13N3O3S268.1156.0∗/113.17021/2316.0Sulfadimethoxine(SDM)C12H14N4O4S311.0156.0∗/108.18031/3819.5Sulfamethizol(SMTZ)C9H10N4O2S2271.0156.1∗/108.06521/368.8Sulfanitran(SNT)C14H13N3O5S336.1294.1∗/198.16519/2221.8Sulfadimethoxine-D6(SDM-D6)#C12H8D6N4O4S317.0156.0852919.5
*Quantitative ion;#Isotope internal standard.
1.4 测定方法
取样品溶液和混合标准溶液各10.0 μL进行测定,以其标准溶液峰的保留时间和MRM两对质谱监测离子对为依据进行定性分析,以定量离子对的峰面积和相应内标物的峰面积的比值为纵坐标,浓度为横坐标绘制工作曲线,以工作曲线比较定量,计算相应待测物的含量。
2 结果与讨论
2.1 色谱条件的优化
QNs结构中含有叔胺基,与色谱填料中的残余硅醇基和金属离子产生氢键或离子交换作用,造成峰形拖尾、展宽,保留时间漂移等现象,该现象成为兽药多残留分析时色谱条件优化的难点[7]。文中所涉及的42种兽药属于中等极性或极性较弱的化合物,选用5款反相色谱柱进行比较:Thermo Hypersil GOLD C18柱(150 mm×2.1 mm,1.9 μm)、Waters Attantis T3柱(100 mm×4.6 mm,3 μm)、Waters Attantis dC18柱(100 mm×2.1 mm,5 μm)、CAPCELL MGⅡ C18柱(150 mm×2.0 mm,5 μm)、Agilent ZORBAX SB-Ag C18柱(150 mm×2.1 mm,3.5 μm)),发现Waters Attantis T3色谱柱可有效克服QNs色谱峰拖尾。这可能是该色谱柱采用专有的键合和封端技术,能够将更多的游离硅羟基反应完全。
用作液-质分析的有机流动相,大都采用甲醇或乙腈,当采用甲醇作为流动相时,有部分极性较弱的药物如萘啶酸、氟甲喹需很长时间才洗脱出来,为缩短分析时间,采用乙腈作为流动相中的有机相。这42种兽药均为[M+H]+的模式,为提高电离效率,增加目标物的响应值,选择在流动相的水相中增加适量甲酸。在实验中发现,随着甲酸浓度增加,各目标分析物的离子对色谱峰响应降低,噪音增大,这可能与药物的准分子离子峰稳定性及离子化效率有关,经比较最终确定流动相体系中甲酸浓度为0.1%。42种药物极性各不相同,与色谱柱的亲和力不同,为更好的将药物分离,采用梯度洗脱方式,针对反相色谱柱,首先用高比例的水相(80%)和低比例的有机相(20%),使极性较强的目标物被洗脱出来,先采用较缓梯度使目标物中的同分异构体,特别是SMT、SMPD和SMM三种同分异构体达到基线分离,接着采用较陡梯度,提高有机相的比例至70%,使极性较弱的目标物尽早出峰,缩短分析时间。在该色谱分离条件下,目标物中具有相同母离子和子离子的6组同分异构体都能得到基线分离,特别是SMT、SMM和SMPD,峰形尖锐、峰宽较窄,各药物保留时间稳定,从3.8 min始至21.9 min结束,出峰分布较均匀,确保了质谱在合适的扫描范围内各色谱峰都有10个以上扫描点数,各目标物得到最佳灵敏度。
在优化的色谱-串联质谱条件下,42种药物总离子流图及6种同分异构体的提取离子流图见图1。
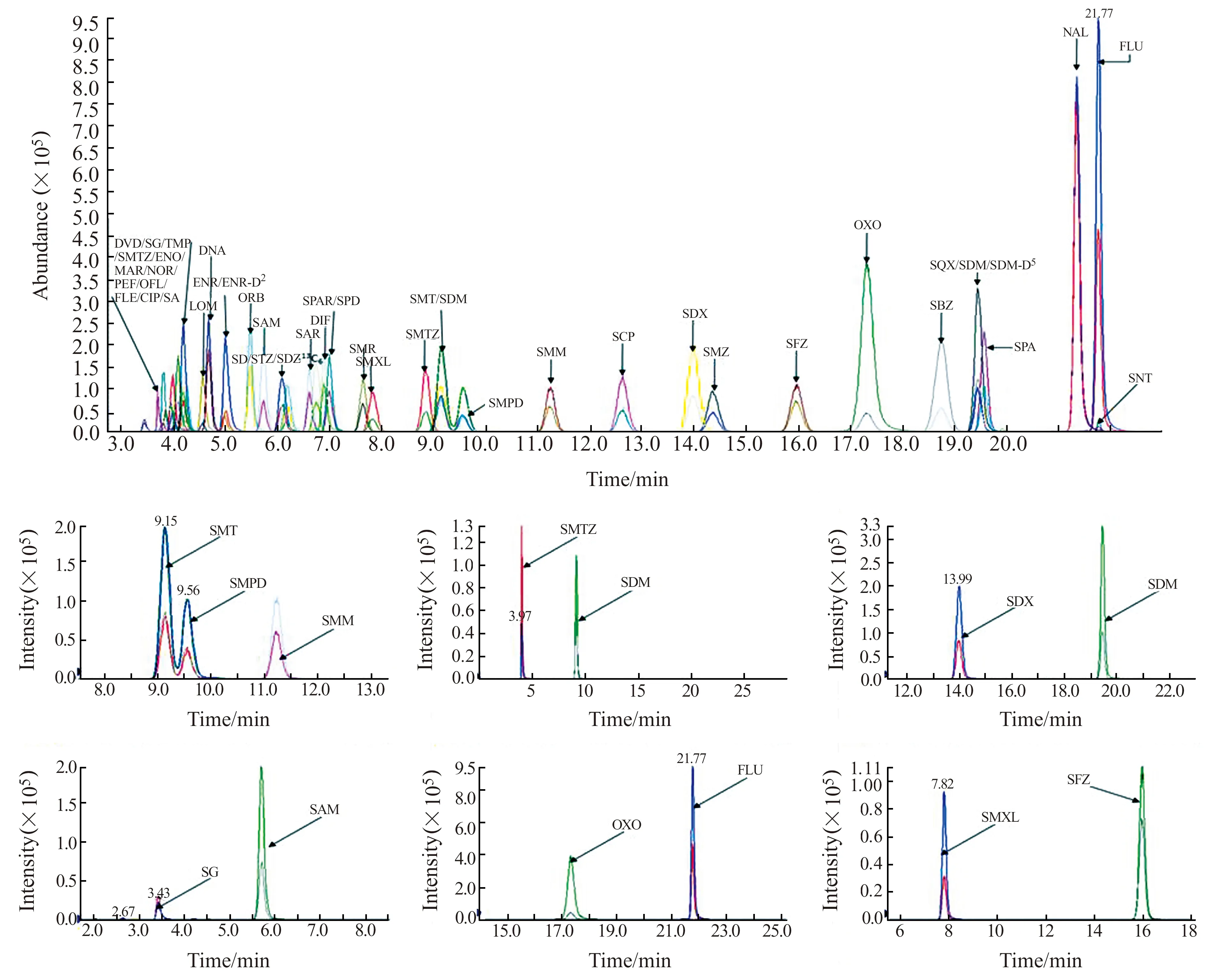
图1 混合基质标准溶液的总离子流(TIC)色谱图和6种同分异构体的提取离子流(EIC)色谱图Fig.1 TIC chromatogram of mixed matrix-standard solution and EIC chromatograms of six isomers
2.2 质谱条件的优化
根据待测物的化学结构,42种待测物均适合在ESI+模式下进行离子化,母离子均为[M+H]+。将42种标准品储备液用甲醇稀释成浓度为1.0 mg/L的标准使用溶液。在ESI+模式下,分别对待测物进行质谱参数优化。先根据药物的相对分子质量通过选择离子监测优化去簇电压(DP)使[M+H]+的响应最大,再以其为母离子,对其子离子进行MS2扫描分析,通过优化碰撞能量(CE)使得子离子的响应最大,挑选灵敏高、干扰小的二对离子,得到MRM离子对及相应质谱采集参数。另外,为了减少样品中杂质对离子源及真空系统的污染,本实验在进入质谱前采用了类似溶剂延迟的功能,色谱柱流出液经六通阀切换至废液中,2.5 min后切换至质谱中采集数据,第23.0 min时六通阀又将柱流出液切换至废液中,这使得极性较强和极性较弱的杂质都被切换至质谱仪之外。
2.3 样品前处理条件的优化
由于乙腈是极性溶剂,可以避免从动物组织中提取出过多的脂肪,同时乙腈具有很好的蛋白沉淀效果和共提物少的特点,可沉淀样品中99%的蛋白,乙腈在兽药残留检测时被广泛运用[1-5]。本方法选择乙腈作为提取溶剂。磺胺类药物有酸碱两性,在不同的溶剂中均有一定的溶解性;喹诺酮类药物在酸性提取溶剂下,更易被萃取。经实验比较,提取液中加入0.1%的甲酸可达到较理想的回收率。
动物源性产品中含大量磷脂,极大干扰目标化合物的分析,降低色谱柱寿命、增大仪器系统的维护成本和难度。本方法采用通过式Oasis PRiME HLB固相萃取(SPE)小柱净化样品,相比保留式SPE净化小柱,无须活化、平衡、上样、洗涤、洗脱步骤,可大幅缩短实验时间,避免在上样及淋洗过程中产生的吸附不完全导致穿透而使目标物损失[8],避免样品堵塞柱子。通过扫描m/z184的母离子,得到近似磷脂类化合物的含量信息。图2比较了经PRiME HLB净化前后的空白猪肉样品的净化效果。说明样品经PRiME HLB净化后,可去除99%磷脂类化合物的干扰。
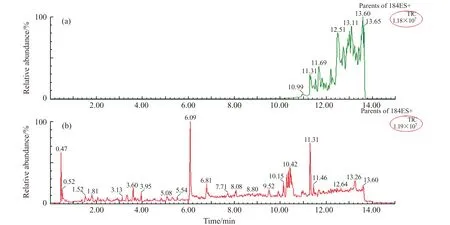
图2 PRiME HLB净化前后的空白猪肉样品效果比较Fig.2 Comparison of blank pork samples before(a) and after(b) PRiME HLB purification
2.4 线性方程与检出限
在选定的色谱分离条件和质谱测定参数下,测定7个水平的基质匹配工作溶液系列,以目标物峰面积与内标物峰面积之比为纵坐标,以目标物的质量浓度(μg/kg)为横坐标做工作曲线,得到线性回归方程,42种药物回归方程的相关系数在0.9978~0.9999之间,表明各药物在相应的浓度范围内呈良好的线性关系。空白样品中添加已知浓度的药物标准品工作液,添加浓度依次降低,按照方法规定的步骤进行检测,以能达到信噪比(S/N)>3时样品的浓度为检测限(LOD),定量离子色谱峰信噪比(S/N)>10为方法定量限,结合药物的最大残留限量(MRL),确定检测限为1.0 μg/kg,定量限为2.0 μg/kg。
2.5 方法的回收率和精密度
取猪肉样品,按2.0、4.0、10.0 μg/kg浓度水平添加标准溶液,每个样品均按本方法做6次平行测定,结果见表2。各药物的平均回收率为81.1%~115.9%,相对标准偏差(RSD)为6.8%~22.1%,符合有关法规的要求。
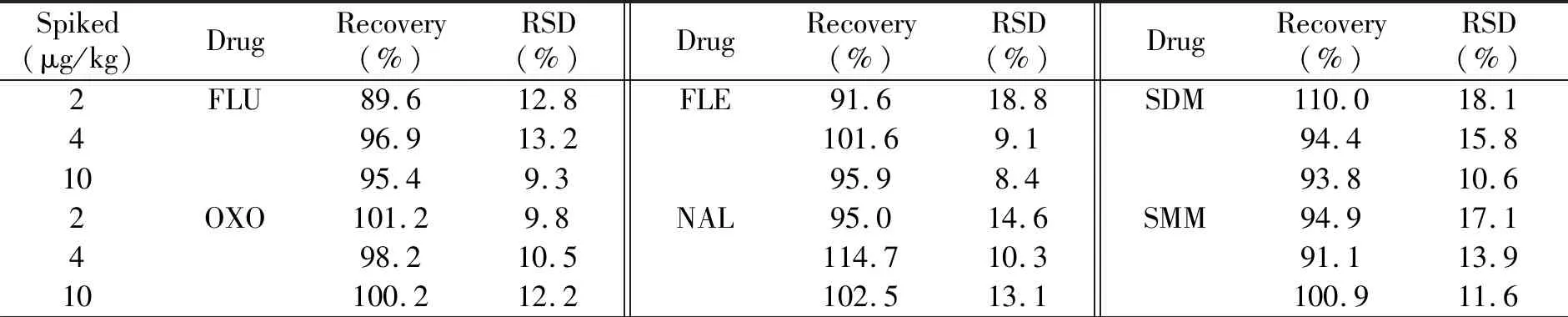
表2 样品中42种药物的回收率和相对标准偏差(n=6)Table 2 Recoveries and RSDs of 42 kinds of drugs in the sample(n=6)
(续表2)
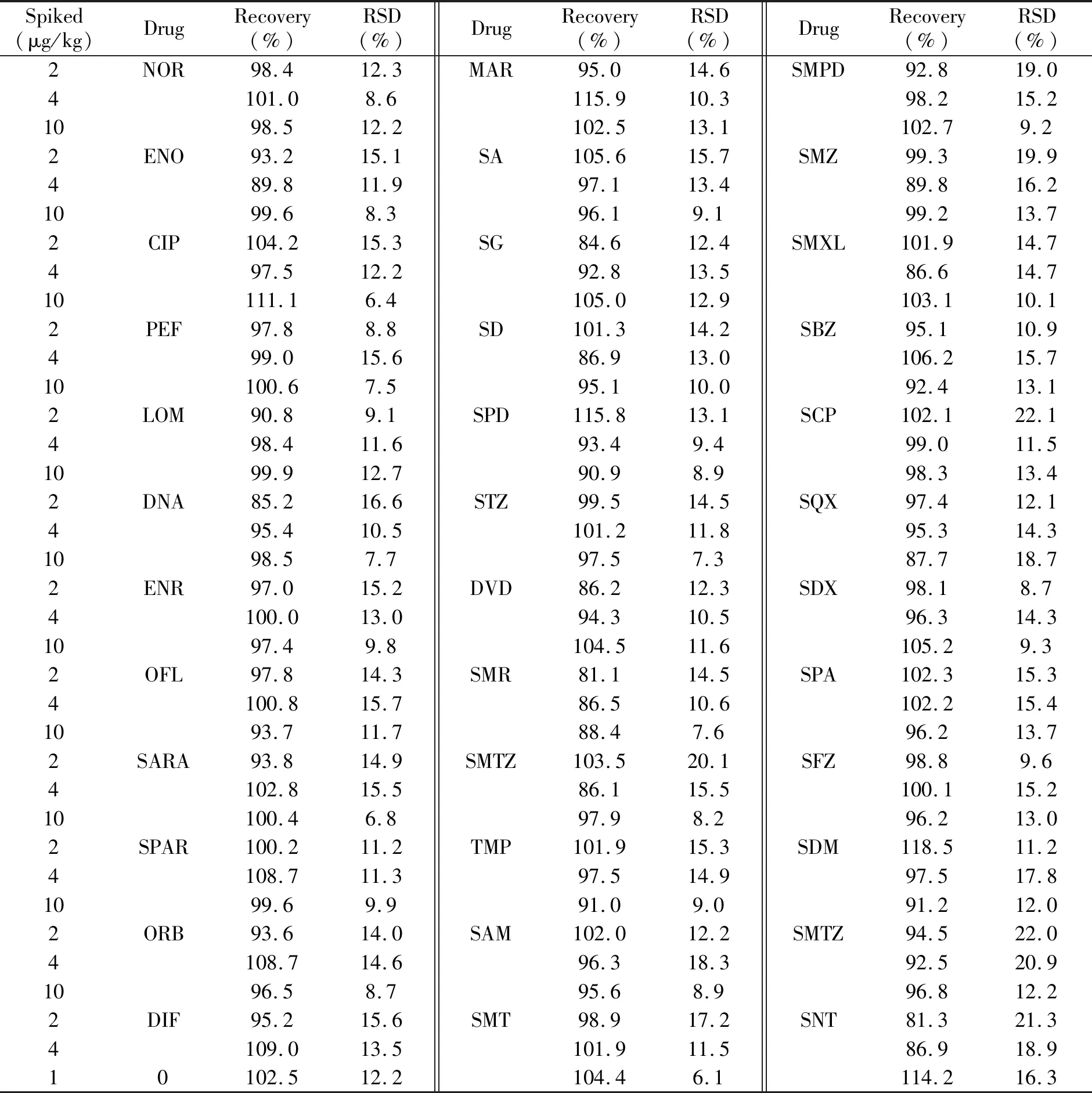
Spiked(μg/kg)DrugRecovery(%)RSD(%)DrugRecovery(%)RSD(%)DrugRecovery(%)RSD(%)2NOR98.412.3MAR95.014.6SMPD92.819.04101.08.6115.910.398.215.21098.512.2102.513.1102.79.22ENO93.215.1SA105.615.7SMZ99.319.9489.811.997.113.489.816.21099.68.396.19.199.213.72CIP104.215.3SG84.612.4SMXL101.914.7497.512.292.813.586.614.710111.16.4105.012.9103.110.12PEF97.88.8SD101.314.2SBZ95.110.9499.015.686.913.0106.215.710100.67.595.110.092.413.12LOM90.89.1SPD115.813.1SCP102.122.1498.411.693.49.499.011.51099.912.790.98.998.313.42DNA85.216.6STZ99.514.5SQX97.412.1495.410.5101.211.895.314.31098.57.797.57.387.718.72ENR97.015.2DVD86.212.3SDX98.18.74100.013.094.310.596.314.31097.49.8104.511.6105.29.32OFL97.814.3SMR81.114.5SPA102.315.34100.815.786.510.6102.215.41093.711.788.47.696.213.72SARA93.814.9SMTZ103.520.1SFZ98.89.64102.815.586.115.5100.115.210100.46.897.98.296.213.02SPAR100.211.2TMP101.915.3SDM118.511.24108.711.397.514.997.517.81099.69.991.09.091.212.02ORB93.614.0SAM102.012.2SMTZ94.522.04108.714.696.318.392.520.91096.58.795.68.996.812.22DIF95.215.6SMT98.917.2SNT81.321.34109.013.5101.911.586.918.910102.512.2104.46.1114.216.3
2.6 与标准方法比较
取已知含量的质控样品,按本法规定步骤检测磺胺嘧啶、磺胺甲恶唑和恩诺沙星,结果见表3。本法的测定值与真实值的含量偏差符合实验室质量控制规范(GB/T27404)《实验室质量控制规范 理化检测》[9]的要求,说明本法与国家标准方法无差异性。

表3 质控样品测定结果(μg/kg)Table 3 Determination results of quality control sample (μg/kg)
3 结论
本实验以虾等动物源性食品为对象,建立了通过式净化-HPLC-MS/MS同时测定动物源性食品中的3类共42种兽药残留的分析方法。经实际样品检测,方法简便、快捷、准确,解决了标准检测方法存大的检测项目少、时间长、前处理繁、成本高等不足,适用于动物源性食品中兽药多残留的快速筛查检测。
