煤间接液化合成油技术研究现状及展望
2020-03-19王学云胡发亭王光耀
王学云,胡发亭,王光耀
(1. 煤炭科学技术研究院有限公司 煤化工分院,北京 100013;2. 煤炭资源开采与洁净利用国家重点实验室,北京 100013;3. 国家能源煤炭高效利用与节能减排技术装备重点实验室,北京 100013)
0 引 言
我国能源资源特点是富煤贫油少气,煤炭资源丰富,储量大,产量高;而石油和天然气储量和产量均较低,严重依赖进口。2018年,我国煤炭探明可采储量达1 145亿t,煤炭产量则达到了36.8亿t,煤炭消费量在我国能源消费总量中占比59%,我国已经成为世界上最大的煤炭生产国和消费国[1-3]。煤炭价格低廉、来源可靠、可清洁利用,因此在未来相当长时期内,煤炭仍将是我国最丰富、可靠、经济的主体基础能源和重要原料。
煤炭间接液化技术是先将煤炭在气化炉内气化得到合成气,合成气通过费托(F-T)合成反应得到分子量分布很宽的液体产物,最后通过对液体产物蒸馏、加氢、重整等精炼过程,得到合格的液体燃料和化学品等[5]。煤间接液化合成油技术生产的燃料油不仅无氮、无硫、无重金属,芳烃含量低,还能副产诸多化学品(如润滑油、烯烃、蜡、含氧化学品等)。因此,煤间接液化技术是促进煤炭高值、高效、高质利用的重要途径,是发挥我国资源优势、保障能源供应安全和稳定供给、促进经济可持续发展的重要战略举措。
本文总结了煤炭间接液化技术的发展历程、研究现状,阐述了国内外在费托合成反应机理、费托合成反应动力学、费托合成催化剂及间接液化工艺等方面的发展现状,并对未来煤炭间接液化技术的研究重点和发展趋势进行了展望,旨在为煤炭间接液化制燃料油及化学品技术的关键技术开发和工业化推广提供理论基础与技术指导。
1 费托合成反应机理
费托合成反应是20世纪20年代初由德国科学家Fischer和Tropsch发现的,因此也被称之为F-T合成反应。费托合成是合成气(CO和H2)在催化剂作用下,通过CO加氢和碳链增长反应,生成液体烃混合物的工艺过程。费托合成反应复杂,包括许多平行反应和顺序反应,相互竞争又相互依存。费托合成的主反应是烷烃生成和烯烃生成,副反应主要是甲烷化、醇类生成、醛类生成、结炭反应等,这些反应发生的概率由工艺条件和催化剂等条件决定。
近几十年,广大科研工作者对费托反应过程进行了广泛深入的研究,提出了几十种反应机理,这些机理是通过设想在费托合成反应中形成含有C、H、O不同中间体的途径或是基于某种模型提出的,虽然在一定程度上能得到试验支持,但截至目前,尚无业内公认的费托合成反应及产物生成机理。
1.1 早期经典费托合成反应机理
早期学者通过同位素示踪、分析催化动力学数据等研究费托合成反应机理,提出了十余种费托合成反应及产物生成机理,但受到较多认可的费托反应机理主要有:CO解离吸附类(如表面碳化物机理)、CO非解离吸附类(如含氧中间体缩聚、CO插入机理等)[4-6]。
1.1.1表面碳化物机理
Fischer和Tropsch针对费托合成反应最早提出了表面碳化物机理。该机理认为在催化剂表面CO首先解离生成活性金属碳化物(CoC、Fe3C2等),其与H2反应生成—CH2—(亚甲基团),—CH2—发生聚合反应生成烷烃和烯烃等。产物中碳链长短取决于氢气活化情况,催化剂表面吸附氢量少时,形成高碳烃;氢气过量则生成甲烷。
表面碳化物机理能较好地解释各种烃类的生成,且在绝大多数Fe系催化剂的费托合成中得到广泛支持。但其无法解释CO生成表面碳化物的速率明显低于液态烃的生成速率,以及支链产物和含氧化合物的生成。
1.1.2含氧中间体(烯醇)缩聚机理

含氧中间体(烯醇)缩聚机理能解释含氧有机物的生成是由于中间体氢化不完全所致,比碳化物机理能更好地说明合成产物分布,以及2-甲基支链和直链产物的生成,但其忽略了表面碳化物在链增长中的作用。
1.1.3CO插入机理
Schulz和Pichler[8]认为,费托合成过程中,被吸附的CO不断插入到金属-烷基键,使链增长形成C—C键,并提出一氧化碳插入机理,认为金属-烷基键是由催化剂表面的亚甲基CH2还原后生成。费托合成反应体系中,首先CO和H2生成甲酰基,甲酰基进一步加氢生成桥式—CH2—,—CH2—加氢和脱水后生成碳烯和甲基,CO不断插入到金属与氢(或烷基)的化学吸附键之间,生成不同长度碳链的烷烃和烯烃产物,反应过程如图1所示。
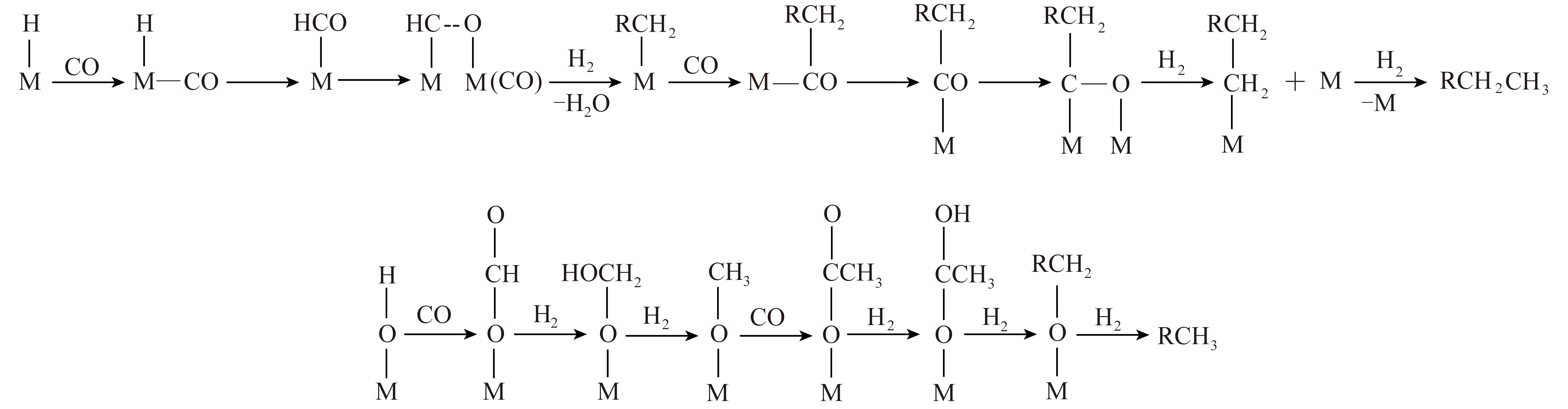
图1 CO插入机理模型
CO插入机理可详细解释直链产物的形成过程以及含氧化合物的形成,但该机理只能依据直链烃和支链烃的相对速率来解释直链烃比支链烃多的原因,目前还无法确定这些基元反应的相对速率。由于该机理对于C—C键的形成只考虑了烷基的转移,较难阐明产物中2-甲基支链的形成过程。
1.1.4双中间体缩聚机理
双中间体缩聚机理(也被称为双活性中心机理)由Nijs和Jacobs[9]提出。该机理既同时考虑了碳化物反应机理和烯醇缩聚反应机理。其核心内容是在费托合成催化剂表面具有2类活性物种,即氢化的氧原子和活化的碳原子。
依据碳化物机理活化碳原子发生烃化反应形成甲烷;根据烯醇缩聚机理通过CO插入实现链增长。双中间体缩聚机理可更好地解释费托合成反应过程中更多的试验现象和反应过程,如甲烷产物不符合Schulz-Flory分布,但其无法解释产物中的支链形成过程。
1.2 近期费托合成反应机理
早期经典的费托合成反应机理仍有许多不足,特别是无法阐明产物分布偏离ASF分布现象。近年来,随着现代表面科学技术在费托合成反应机理研究中的应用,在费托合成反应过程中催化剂表面组成变化、不同碳数增长、吸附物种的行为和产物分布等均取得了研究进展。由此提出了C2活性物种机理、烯烃再吸附的碳化物机理以及网络反应机理等[10-12],较为理想地解释了费托合成反应中出现的特殊产物分布现象,是早期经典费托合成机理的有益补充。
目前,许多学者是从反应的宏观行为或催化剂性质对费托合成反应机理开展相关研究和推测,对真实反应机理尚未完全阐明。采用原位表征、高能同步辐射、量子化学、密度泛函理论计算等研究催化剂活性中心,是揭示费托反应反应机理本质的重要途径,也是未来费托反应机理基础研究的方向。因此,清晰认识费托合成反应机理对于高活性、高选择性催化剂的定向设计以及反应产物的精准调控至关重要,对新一代高活性费托催化剂和费托合成新工艺的开发具有重要的指导作用。
1.2.1C2活性物种机理
费托反应过程中,CO和H2在催化剂表面的吸附、CO在催化剂表面的解离和金属表面上碳和氢的作用等是费托反应的关键步骤。CO在催化剂表面解离后,形成活性碳原子,活性碳原子和氢气发生氢化反应形成—CH2—,—CH2—聚合促进了碳链增长,是烃类产物形成的先决条件。Mims等[13]利用13C示踪法研究了费托合成在Ru、Fe、Co催化剂上碳氢化合物的增长机理,发现C2本体是链引发物种,C2链的形成过程较慢,催化剂表面逐渐形成C2中间体(亚乙烯基、亚乙基),形成后的中间体活跃,能迅速与其他引发物发生链增长反应。
1.2.2烯烃重吸附的碳化物机理
基于费托反应中烯烃作为链引发物种重新吸附到催化剂表面的现象,学者们从烯烃在费托反应中的插入、异构化、烯烃再吸附等方面进行了研究,结果表明[12],C2H4的再吸附和强反应性使油和氧化物含量增加,说明烯烃作为费托合成反应产物会重新在催化剂表面吸附,并再次参与费托合成反应,从而发现了烯烃对产物分布的影响效应。
Bell等[16]根据烯烃在费托反应中的影响规律,将烯烃重吸附机理进行归纳:① 表面C2物种为链引发物种,C2H4的加入会链入反应碳链中,促使C3+反应速率增加;② C2H4不会裂化为CH4产物;③ 解离吸附后CO形成的活性碳物种与氢气反应生成亚甲基;④ C2H4的加入不影响表面的聚合和CO氢化步骤。
基于碳化物机理的烯烃再吸附理论,得到了大量试验结果证实,认为在催化剂表面的烯烃重新吸附,一部分是作为链引发和链增长物种,另一部分会通过氢化、插入、再吸附等反应,与H2和CO发生链形成反应,生成更多碳数的费托合成烃类产物。目前,对烯烃的重吸附是否影响CH4生成速率仍没有定论。
1.2.3网络反应机理
费托反应机理研究过程中发现,不同反应途径产生不同的活性中间体,进而形成了不同的产物。美国加州大学的Rofer-Depoorter[17]提出了基元反应间相互交叉的网络机理模型,并引入零有效速率的概念,将对反应机理的研究转变为对基元反应速率的对比研究。网络反应机理认为,F-T合成反应包括反应物吸附、吸附态中间体相互作用、氢化和链增长、产物生成等4个阶段,是一组基元反应形成的复杂网络体系。在不同反应条件下,各基元反应均可能存在,其速率不同。虽然网络反应机理在化学本质上没有新突破,但其通过表面分析技术和计算机软件模拟阐明反应器中发生的实际反应过程,为费托合成反应机理的研究提出了新思路。
2 费托合成反应动力学
费托合成是一个包含许多顺序和平行反应、多种机理并存的复杂系统,由于其反应复杂、产物碳数分布较宽、存在大量变数、反应机理不确定等原因,至今仍未完全阐明费托合成反应动力学规律及反应历程。在不同反应器中,利用不同的合成催化剂、在不同的操作条件下,对费托合成反应动力学进行了大量研究,获得了多种表达式和动力学方程。费托合成反应动力学的研究分为描述CO消耗速率动力学模型和详细动力学模型两大类。
2.1 CO消耗速率动力学模型
Anderson等[19]由含氧中间体缩聚机理推导出LHHW动力学模型,模型中H2的反应级数为一级,H2和CO占据催化剂的表面活性位,其中H2O对合成反应有抑制作用。Anderson的机理型动力学模型成为许多学者进行费托合成反应动力学研究的基础,基于不同的催化剂、反应器等衍生出多种LHHW型动力学模型。Deckwer等[20]发现,当H2/CO<0.8或H2O浓度比较低时,需采用不同的动力学模型。Yates等[21]在浆态床反应器中,采用铁基催化剂,在CO2与合成气同时进料的条件下研究了费托合成动力学,未发现CO2对反应存在抑制作用。Van Steen等[22]假定费托合成反应为不可逆的氢化反应,得到的动力学模型中分母为二次方,为了强调“空位”的概念,在动力学方程分母中引入了常数项,并发现H2O是通过影响表面碳的中间体浓度从而影响动力学。Laan等[23]采用统计学方法,推导出多种LHHW型费托合成动力学模型,认为动力学表达式中有必要加入空位,因此其推导的LHHW型模型均考虑了表面空位的影响。吉媛媛等[24-25]在碳化物机理的基础上建立了烃生成基元步骤,根据甲酸盐机理建立了超细Fe-Mn催化剂的费托合成中CO消耗速率动力学模型,模型中包含了CO2生成的基元反应步骤。鲁丰乐等[26]采用Co/SiO2和Fe-Cu-K/SiO2催化剂,在等温积分反应器研究了费托合成的本证动力学,推导出对试验数据拟合较好的LHHW型动力学方程。
目前,大多数学者对费托合成反应动力学的研究均采用CO消耗速率动力学模型,但该动力学模型一般是采用经验的幂指数型动力学表达式,或依据特定的反应机理及速率控制步骤获得LHHW型动力学表达式,虽能较好地预测CO消耗速率,但无法提供费托合成产物分布的详细信息[27]。目前仍没有模型可适合各种反应条件下的反应动力学行为,且模型动力学方程带有一定的经验性。
2.2 详细动力学模型
CO消耗速率动力学模型无法预测费托合成产物分布,无法满足反应过程开发和反应器设计的工程实践需要。详细动力学模型可涉及链引发、链增长和链终止等费托合成复杂聚合机理的详细细节,可同时预测CO或合成气的消耗速率以及费托合成产物的详细分布。
Lox等[28]在经典的碳化物机理基础上,建立了沉淀铁催化剂的费托合成详细动力学模型,该模型给出了产物分布随操作条件的变化关系,能提供预测各烃选择性的信息。但由于该模型依据理想的ASF聚合机理,对于产物分布会产生一定的偏差。马文平等[29-30]考虑到费托合成中的烯烃再吸附,推导出Fe基催化剂费托合成产物分布的详细动力学模型,认为碳链增长因子与碳链长度相关,而非ASF机理所描述的常数。
常杰等[31]以亚甲基生成步骤为速率控制步骤,并考虑在浆液中产物的溶解度随碳链长度变化的关系以及烯烃的再吸附和二次反应,推导出一系列浆态反应器中Fe-Cu-K/SiO2催化剂的费托合成详细动力学模型,经参数回归和模型检验,得到最优的费托合成详细动力学模型。腾波涛等[32]基于CO插入机理和亚甲基插入机理,综合考虑费托合成反应中烯烃、醇与酸的再吸附与二次反应以及非本征效应对产物分布的影响,建立了综合的费托合成详细动力学模型,得到各产物的生成速率表达式。常杰等[31]在深入认识费托合成中关键基元化学反应步骤的基础上建立的最优详细动力学模型,可较完整准确地实现对 Fe-Cu-K-SiO2催化剂上烃生成行为的描述。滕波涛等[32]利用无梯度反应器内温度、压力及气氛均一的优势,分离出烃、含氧化合物生成及水煤气变换反应,单独建立优化的动力学模型,进而得到综合动力学模型,该模型从不同层次描述了费托合成反应行为。
在推导详细动力学模型的表达式过程中,需要考虑碳链增长因子,并认为其与碳数有关,使详细动力学模型的推导和计算过程较复杂。
3 费托合成催化剂
费托合成反应只有在催化剂作用下才能实现,催化剂对反应速率、产品收率及分布、原料转化率以及工艺条件等均具有决定性影响。元素周期表中的第Ⅷ族过渡金属元素对费托合成反应均具有催化活性,其中钴(Co)、铁(Fe)、镍(Ni)、钌(Ru)是最活泼的金属元素[33]。
金属Ru是活性最高的费托合成催化剂,具有链增长能力强、反应温度低等优点,但地球储量少、价格高限制了其工业应用,目前仅具有理论研究价值。Ni尽管也具有较高的CO加氢活性,但甲烷化反应严重,不利于费托合成制备液体燃料,也逐渐被弃用。由于Fe基和Co基催化剂加氢活性好,来源广泛,储量丰富,是目前工业上普遍应用的费托合成催化剂。
3.1 铁基催化剂
铁基催化剂适用于较宽泛的反应条件范围,具有更好的耐硫性,储量丰富,价格低廉,因此尽管碳链增长能力相对较弱、副产物CO2选择性较高,仍在制备低碳烯烃上得到广泛应用。工业用铁催化剂主要有沉淀铁催化剂、熔铁催化剂和烧结铁催化剂,沉淀铁催化剂用于固定床和浆态床反应器,熔铁和烧结铁催化剂用于流化床反应器。
目前铁基催化剂活性相、助剂影响规律、催化机理、活化机理以及CO2形成机理等研究取得了重要进展,为铁基催化剂活性的提高、制备工艺的优化、新型催化剂的开发奠定了坚实的试验和理论基础。
Fe基催化剂在还原、活化和反应过程中,相态结构不断变化。Fe基催化剂经预处理后,α-Fe2O3相态首先还原为Fe3O4,之后在不同反应条件下部分转化为碳化铁及α-Fe物种,预处理条件决定了最终的相态种类[34]。χ-Fe5C2、Fe7C3、θ-Fe3C、ε-Fe2C等是报道较多的碳化铁物种,目前由于Fe基催化剂的物相组成复杂,准确鉴别活性相种类非常困难,普遍认为Fe基催化剂的主要活性相是χ-Fe5C2[35]。
铁基催化剂中常添加K作为助剂,Mahyuddin等[36]研究了K、Na助剂的作用,发现助剂促进了CO的吸附,K比Na具有更强的促进作用,原因是碱金属助剂属于电子型助剂,能加强催化剂与反应物之间的相互作用,电子效应大小取决于碱性强弱。近年来,采用计算机技术的密度泛函理论(DFT)计算化学在铁基催化剂领域得到广泛应用,在Fe基催化剂反应机理的理论研究方面取得了显著进展。CO在Fe催化剂表面进行化学吸附,Fe—C成键,O原子与Fe不直接成键;Fe催化剂的不同相态和不同晶面CO活化路径不同[37]。Petersen等[38]发现,Fe(110)面的主要活化方式是H辅助CO解离路径,χ-Fe5C2(010)和(001)面CO最稳定的吸附位点为atop位和4-fold位。Pham等[39]采用DFT计算化学研究了χ-Fe5C2表面CH4形成和C—C偶联反应,发现其链增长是通过C—C偶联进行,而C+CH和CH+CH是能垒最小的路径。
3.2 钴基催化剂
钴基催化剂具有较高的CO加氢活性和较强的链增长能力,产物以长链烃为主,不易积碳和中毒,对水煤气变换反应不敏感,应用广泛。但工业应用中,钴基催化剂存在成本高、甲烷生成量大、副产低压蒸汽量偏大等问题,在提高催化剂使用寿命、降低甲烷选择性等方面需完善。为此,学者们对钴基费托合成催化剂进行大量研究,如制备方法优化、加入助剂、调控物相结构以及探索反应温度、压力、反应气氛等,以期提高其催化性能,对钴基费托合成反应的工业化生产具有重要意义。
费托合成反应过程中钴基催化剂发生复杂的相态转变。Senecal等[40]发现,经H2预处理和合成气(H2+CO)处理后,Co催化剂的相态变化过程为:Co3O4→CoO→Co,且在高温条件下还可能形成CoxCy。研究发现[41-42],Co催化剂具有面心立方fcc-Co和六角密堆hcp-Co两种不同的晶型结构,hcp-Co晶型结构更易形成,但晶体粒径小于100 nm时,fcc-Co晶型更稳定;反应温度大于695 K时,Co催化剂相态会从hcp-Co转变成fcc-Co,因此低温费托合成中仅存在hcp-Co。Lyu等[43]对fcc-Co和hcp-Co两种晶体结构催化剂进行了评价,发现hcp-Co表现出更高的碳氢物种选择性及CO加氢活性。Qin等[44]发现,fcc-Co(001)面在235 ℃左右时甲烷选择性较高,(111)和(112)晶面的产物具有较高的烯烷比。
采用计算化学等对钴催化剂反应机理进行研究,从微观层面揭示反应本质是目前研究热点,这些机理在一定程度上得到了费托合成试验的验证。Liu等[45]发现,Co(001)表面OH辅助的解离路径热力学及动力学优先,由于OH表面覆盖度低,因此H辅助解离形成C1氧化物中间体的路径是主要的CO解离路径。Zhang等[46]发现,在钴催化剂(1010)表面,CHO物种的解离是CH物种的主要来源,CH2O物种的直接解离和H辅助解离是CH2和CH3物种的主要来源;而钴(1011)表面C和CH物种最多,且主要来源于CO直接解离和C物种的逐步加氢。在费托合成反应过程中,催化剂多个相态并存,Pei等[47]通过试验研究发现,Co2C仅在反应温度493 K左右时部分分解为Co,Co-Co2C界面催化剂比单纯Co2C界面具有更高的C2+碳氧化合物选择性。
钴催化剂在提高费托合成中间馏分油的选择性方面具有优异性能,因此Sasol和Shell公司开发了新一代钴催化剂,主要成分为Co-ZrO2-SiO2。有文献报道[11],制备的添加贵金属的钴催化剂,在225 ℃下,反应产物全部为烃类;另外,用Co(CO)8和负载Pt或Pd的载体制成的催化剂用于浆态床合成等。
3.3 其他新型催化剂
虽然工业催化剂的水平已有较大提高,但新型催化剂的研究与开发仍在持续进行,并取得了显著进展。
1)复合型双功能催化剂
复合型双功能催化剂(如双金属催化剂或金属-分子筛双功能催化剂)的出现是费托合成催化剂研究取得的重要进展。双金属的协同作用可提高催化剂的活性、稳定性和分散性,金属-分子筛催化剂既具有活性金属的链增长能力,又具有分子筛的酸催化特点。复合型催化剂不仅选择性高,且保持了较高的反应速率,性能优异,但卤化物污染、积碳、相变、烧结均会造成复合型催化剂中毒和失活。由于复合型催化剂属于新型催化剂,尚未在中试或工业装置上应用,缺乏长周期寿命验证试验,因此有必要进行复合型催化剂的寿命研究。
马利海等[48]利用水热方法法制备了Fe-Mn双金属催化剂,发现Fe-Mn催化剂具有较高的CO加氢活性,在对Fe-Mn催化剂进行钾改性后,低碳烯烃的选择性显著提高。程世林[12]以Co/SiO2为硅源,采用无溶剂原位合成方法制备了钴/分子筛复合型催化剂Co/ZSM-5,试验结果表明,72 h晶化的复合型催化剂具有最高的汽油选择性,达72.3%。
2)多元金属催化剂
多元金属催化剂可改善催化剂的活性、选择性和使用寿命。在费托合成催化剂中加入对CO有较强吸收能力的Mn、V、Ti等金属,可提高对低碳烯烃的选择性,加入卤素化合物(如KCl或KBr),也有较好效果[49]。
罗伟等[50]利用共沉淀方法制备出Co-Fe-Mn多元金属催化剂,发现添加Fe可改善催化剂的孔结构和分散性,添加Mn可促进多中心活性吸附位的形成和活性组分的分散。李江兵[51]采用共沉淀-浸渍-焙烧方法,制备出多元金属催化剂FeMnK和FeMnNa,并考察了不同金属含量的影响,结果表明,增加Mn含量,可提高烯烃的链增长几率,增加Na和K含量可促进高碳烯烃和烷烃的生成。
3)新型载体催化剂
近年来,学者对载体性质及其对催化性能的影响规律进行研究,新型载体材料如碳纳米管、石墨烯、分子筛、活性炭等被引入费托合成催化领域,促进了新型负载型催化剂的涌现,提高了费托合成技术的发展。
碳纳米管的导电性、机械强度、热稳定性良好;石墨烯层状结构,层间距较大,导电、导热性好且易被修饰。Schulte等[52]研究发现,碳纳米管负载的Fe基催化剂对烯烃具有较高的选择性和低链增长概率。Chen等[53]以氮掺杂的石墨烯作为载体制备了铁基催化剂,低碳烯烃的选择性在50%左右。
4 煤间接液化工艺
4.1 发展概况
煤炭间接液化工艺主要包括煤气化、煤气净化、费托合成反应以及产物分离精制等过程,反应器、催化剂、操作条件以及产品方案不同,间接液化工艺技术不同。
目前,国外实现工业化的间接液化工艺有南非Sasol公司的费托合成技术、荷兰Shell公司的SDMS工艺以及美国Mobil公司开发的MTG(Methanol to Gasoline)甲醇生产汽油合成技术[6,11]。近年来,国外相关公司也开发了许多煤间接液化工艺技术,如丹麦Topsoe公司的Tigas工艺、美国Exxon公司开发的AGC-21工艺以及Mobil公司开发的STG技术等[11,51],虽然这些工艺技术先进,但都未实现商业化应用。
我国于20世纪50年代开始对费托合成进行初步研究,但由于大庆油田的发现,费托合成研究中断了30年。20世纪80年代,由于经济快速发展使油品需求量增大,国内相关科研单位和企业重新开始对费托合成进行研究[51,54]。中科院山西煤化所开发了铁基、钴基催化剂和超细粒子铁、锰催化剂以及浆态床反应器,形成了多种煤间接液化工艺,并实现了百万吨级产业化。兖矿集团开发出新型催化剂、固定床和流化床反应器,形成了成套的高低温费托合成工艺技术,并实现了百万吨级产业化。
目前工业上应用的煤间接液化工艺众多,按照费托合成温度,可划分为低温间接液化和高温间接液化工艺。低温费托合成工艺操作条件易控制、油收率高,但其柴油产品密度低、反应器内传质阻力较大、副产的低品位蒸汽难以利用,导致能量转化效率低。因此,开发高活性催化剂、优化反应器设计、加强低品位蒸汽的高效利用是低温煤间接液化工艺未来发展的方向。高温煤间接液化油品质量好,产品中高附加值烯烃含量高,能量转化效率高,但甲烷选择性高、油品收率低、催化剂易碳化。研制低甲烷选择性、良好抗磨性能的催化剂,设计和控制适合高温运行的反应器是高温煤间接液化工艺优化和完善的技术关键。
4.2 国内煤间接液化工艺
4.2.1中科院山西煤化所间接液化制油工艺
中科院山西煤化所从20世纪80年代开始进行煤炭间接液化技术研究及工程开发,开发出高活性的铁基、钴基费托合成催化剂[55],并在催化剂研发、中试装置验证的基础上,开发了固定床间接液化工艺(MFT)以及浆态床和固定床结合的两段间接液化工艺(SMFT)[55-57]。
MFT间接液化工艺将传统费托合成与沸石分子筛相结合。该工艺中,净化处理后的合成气,先进入一段反应器中与铁基催化剂作用,生成C1~C40的宽馏分烃类产物,之后进入装有择形分子筛催化剂的二段反应器中,再进行烃类催化转化反应,最终转化为C5~C11的汽油馏分。
SMFT间接液化工艺是浆态床-固定床两段法工艺。该工艺中,合成气首先进入装有超细粒径铁基催化剂的反应器中,获得烃类产物,烃类产物在ZSM-5分子筛上转化为高辛烷值汽油。SMFT工艺显著提高了费托合成过程的效率和液体燃料组分的收率。
2006年,山西煤化所和其他企业联合成立了中科合成油技术有限公司。2008年,中科合成油公司研制出高温浆态床费托合成铁基催化剂,并进行了中试装置试验验证和工艺条件优化,形成了成熟的高温浆态床合成油工艺技术。2009年,利用高温浆态床费托合成工艺,分别为山西潞安集团、内蒙古伊泰集团、神华集团建成了3套16万~18万t/a的工业示范装置。2016年,神华宁煤400万t/a、内蒙古伊泰120万t/a、山西潞安100万t/a的煤间接液化工业示范装置建成投产,实现了中科合成油公司的高温浆态床费托合成工艺的百万吨级工业化应用,达到国际领先水平[33,58]。
4.2.2兖矿集团的煤间接液化制油工艺
兖矿集团从1998 年开始进行煤间接液化制油技术的基础研究和工艺开发,研发重点为低温铁基浆态床技术和高温铁基固定流化床技术。2002年,兖矿集团组建了上海兖矿能源科技研发有限公司(简称为兖矿能源公司),从事煤间接液化技术的开发及工程化[59]。
兖矿能源公司研制了低温费托合成铁基催化剂,研发了煤间接液化制油全过程模拟软件和三相浆态床反应器[60-61]。在此基础上,建成了百吨级催化剂中试装置和千吨级费托合成中试装置,获得了优化的工艺操作条件和工程设计基础数据,形成了低温浆态床煤间接液化工艺(图2)。该工艺采用铁基催化剂和三相浆态床反应器,工艺流程主要包括催化剂前处理、费托合成及产品分离3部分。2015年,兖矿榆林100万t/a的煤间接液化工业示范项目建成投产,采用兖矿能源公司研发的低温浆态床间接液化成套工艺,该项目至今运转情况良好[60]。
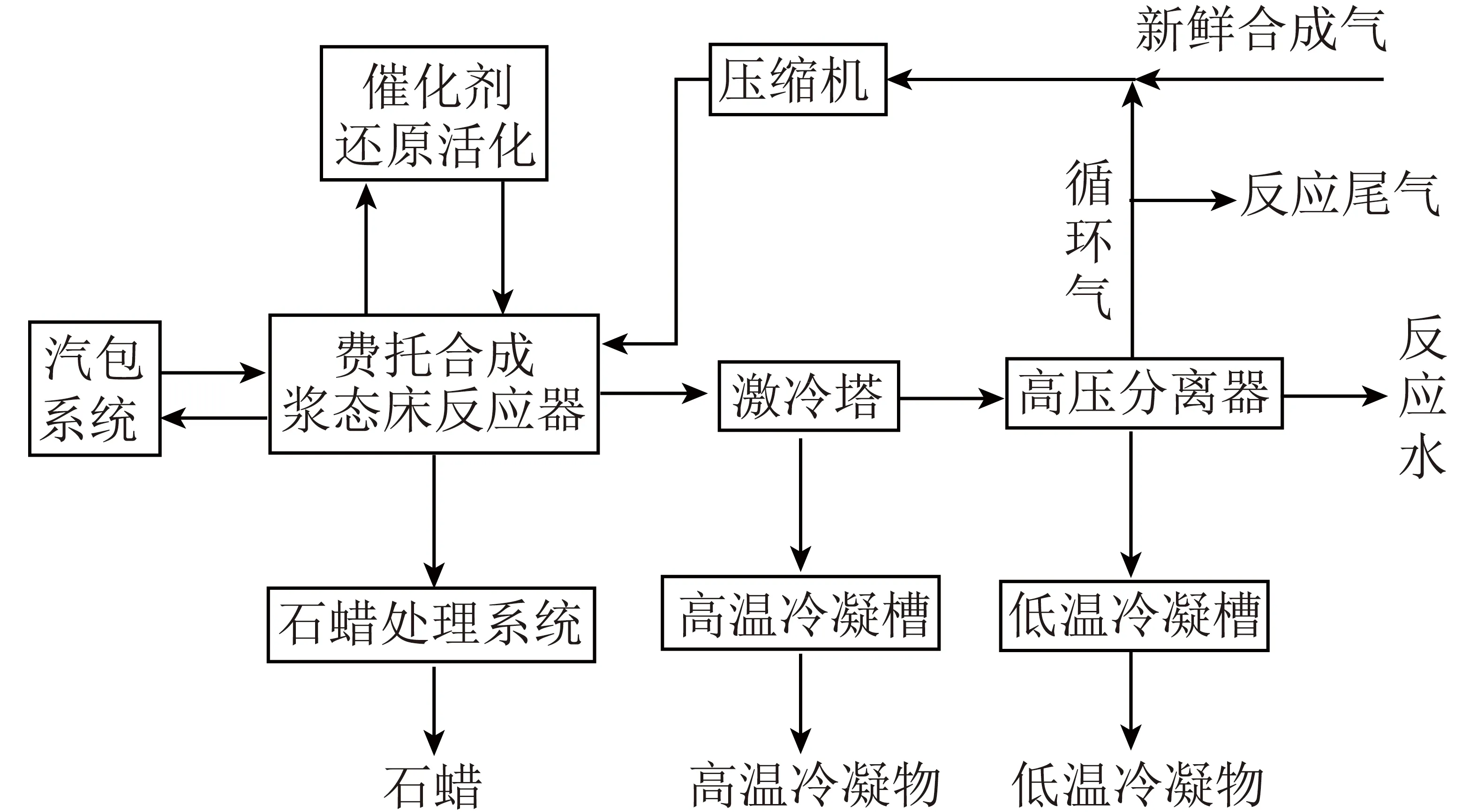
图2 兖矿低温煤间接液化工艺流程
兖矿能源公司[59-61]还开发了高温煤间接液化工艺(图3)。该工艺中,净化的合成气进入固定流化床反应器,在340~360 ℃条件下与沉淀铁催化剂作用,进行费托合成反应。费托合成生成烃类化合物,经冷却、分离、加氢提质后得到汽油、柴油等产品。该工艺轻质烃产率较高,并经过了千吨级的中试装置工艺验证和长周期考察。
5 结语及展望
在我国石油、天然气对外依存度再创新高的背景下,结合我国化石能源特点及现状,加强煤间接液化技术的相关基础研究,积极研发先进的依托费托合成技术的煤间接液化工艺生产汽油、柴油等石油产品和化工产品,从而提高煤炭利用效率,符合我国大力提倡使用清洁能源和建设环境友好型社会的发展需求,具有很大的市场价值。
1)费托反应机理复杂,产物众多,使现有的机理模型均存在一定的片面性。由于研究手段的限制,目前对费托合成反应机理研究是基于催化剂表面或反应的宏观表现获得的信息,无法深入费托反应本质。借助于原位分析表征和表面科学技术的进步,未来从催化剂活性中心角度研究费托合成反应行为,并从几种机理综合考虑来阐释费托反应本质是一种有意义的尝试。
2)严格复杂机理的费托合成详细动力学在工程设计与分析中具有重大作用,使其成为当前的研究热点,但详细动力学研究面临的问题除了需要建立高精度的模型外,还需要有对费托合成复杂产物的详细准确分析。因此,准确定量包括高碳数蜡在内的烃类产物组分、解决低碳烃等组分在高碳烃蜡中的溶解问题,是今后费托合成反应详细机理动力学研究的攻关方向。
3)目前,在对铁基和钴基费托合成催化剂的开发过程中,对K助剂与CO2选择性的影响研究较少,应加强K助剂对CO2形成机理的影响研究。Mn作为费托合成催化剂的主要活性成分之一,其潜在价值尚未得到充分发挥,应予以重视。
4)工业化应用中常见的F-T合成反应器有固定床、流化床(循环流化床和固定流化床)、浆态床等,其中浆态床反应具有温度分布均匀、操作条件易于调控、可在线更换催化剂等优势而受到重视。
5)由于高温费托合成技术和低温费托合成技术在产品结构方面均有局限性,因此开发高温与低温费托合成工艺联产系统,可充分集成低温费托合成和高温费托合成技术在工艺和产品方面的优点,提高燃料品质,产品方案更加灵活,进而提高企业抵抗市场价格波动的能力,具有显著的经济效益和社会效益。
