醒脑颗粒质量标准研究
2020-03-15马冬云
姜 燕,马冬云
(江苏省连云港市食品药品检验检测中心,江苏 连云港 222006)
醒脑颗粒为江苏省连云港市中医院制备的医院制剂,主要由胆南星、石菖蒲、白蒺藜、玄参、黄芩、生大黄、玉竹、冰片等中药组方,临床用于治疗中风(中脏腑)急性期、风痰火壅于清窍所致神昏、痰涌、腹实不通等,收载于《江苏省食品药品监督管理局医疗机构制剂标准》[1]。本研究中采用薄层色谱(TLC)法对制剂中的黄芩、冰片、大黄、玄参进行定性鉴别[2-5],采用高效液相色谱(HPLC)法测定制剂中黄芩苷、大黄素、大黄酚含量[6-9],为其质量控制提供参考。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260 型高效液相色谱仪(美国安捷伦科技有限公司);BP211D 型电子分析天平(北京赛多利斯天平有限公司,十万分之一);KQ-500TDB 型高频数控超声波清洗仪(昆山市超声仪器有限公司,功率为120 W,频率为40 kHz);Linomat 型薄层点样仪(瑞士卡玛公司)。
1.2 试药
黄芩苷对照品(批号为110715-201720,纯度为93.5%),冰片对照品(批号为110743-200905,纯度为98.0%),哈巴俄苷对照品(批号为111730-201106,纯度为96.0%),大黄素对照品(批号为110756-201512,纯度为98.7% ),大黄酚对照品(批号为110796-201621,纯度为99.2% ),大黄对照药材(批号为120902-201010),均购自中国食品药品检定研究院;醒脑颗粒(江苏省连云港市中医院制剂室自制,批号分别为20180319,20180110,20180523);乙腈、甲醇为色谱纯,水为超纯水。
2 方法与结果
2.1 TLC 鉴别
黄芩:取样品适量,研细,取约4 g,加甲醇15 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液。另取黄芩苷对照品,加甲醇制成每1 mL 含1 mg 的溶液,作为对照品溶液。取阴性样品4 g,同法制备阴性对照品溶液。精密吸取上述3 种溶液各6 μL,分别点于同一硅胶G 薄层板上,以乙酸乙酯-甲酸-水(9 ∶1 ∶1,V/ V/ V)为展开剂,展开,取出,晾干,喷以2%三氯化铁乙醇溶液。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰。详见图1 A。
冰片:取样品适量,研细,取约0.3 g,加石油醚(30 ~60 ℃)5 mL,超声处理10 min,滤过,取滤液作为供试品溶液。另取冰片对照品,加石油醚(30 ~60 ℃)制成每1 mL 含1 mg 的溶液,作为对照品溶液。取阴性样品0.3 g,精密称定,按供试品溶液制备方法制备阴性对照品溶液。精密吸取对照品溶液5 μL、供试品溶液及阴性对照品溶液各15 μL,分别点于同一硅胶G 薄层板上,以二氯甲烷为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在100 ℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰。详见图1 B。
大黄:取样品适量,研细,取约2 g,加甲醇20 mL,超声处理15 min,滤过,滤液蒸干,残渣加水10 mL 使溶解,再加盐酸溶液1 mL,置水浴中加热30 min,立即冷却,用乙醚振摇提取2 次,每次10 mL,合并乙醚液,蒸干,残渣加三氯甲烷1 mL 使溶解,作为供试品溶液。另取大黄对照药材0.1 g,同法制成对照药材溶液。取阴性样品2 g,同法制成阴性对照品溶液。精密吸吸取上述3 种溶液各6 μL,分别点于同一硅胶G 薄层板上,以石油醚(30 ~60 ℃)-甲酸乙酯-甲酸(15 ∶5 ∶1,V/ V/ V)的上层溶液为展开剂,展开,取出,晾干,置紫外光(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同的5 个橙黄色荧光主斑点,阴性对照无干扰。详见图1 C。
玄参:取样品适量,研细,取约3 g,加甲醇25 mL,浸泡1 h,超声处理30 min,滤过,滤液蒸干,残渣加水25 mL 使溶解,用水饱和的正丁醇振摇提取2 次,每次30 mL,合并正丁醇液,蒸干,残渣加甲醇5 mL 使溶解,作为供试品溶液。取哈巴俄苷对照品,加甲醇制成每1 mL含1 mg 的溶液,作为对照品溶液。取阴性样品3 g,同法制备阴性对照品溶液。精密吸取上述3 种溶液各10 μL,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇-水(12 ∶4 ∶1,V/ V/ V)的下层溶液为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,热风吹至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰。详见图1 D。

图1 薄层色谱图
2.2 黄芩苷含量测定
2.2.1 色谱条件
色谱柱:AgilentEclipseXDB-C18柱(150mm×4.6mm,5 μm);流动相:甲醇-0.4%磷酸溶液(47 ∶53,V/ V);流速:1.0 mL/min;柱温:30 ℃;检测波长:280 nm;进样量:10 μL。
2.2.2 溶液制备
取黄芩苷对照品20.72 mg,精密称定,加70%乙醇溶解制成质量浓度为207.2 μg/mL 的对照品贮备液。精密量取贮备液适量,加70%乙醇制成质量浓度为51.8 μg/mL 的对照品溶液。取样品适量,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加入70%乙醇50 mL,称定质量,超声处理1 h,放冷,再称定质量,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液,作为供试品溶液。按处方比例和工艺制备不含黄芩饮片的阴性样品,并按供试品溶液制备方法制备阴性对照品溶液。
2.2.3 方法学考察
专属性试验:取上述3 种溶液各10 μL,按拟订色谱条件进样测定,记录色谱图。结果见图2,阴性对照无干扰,表明方法专属性良好。
线性关系考察:精密吸取对照品贮备液0.5,1.0,2.0,3.0,4.0 mL,分别置10 mL 容量瓶中,用70%乙醇稀释至刻度,摇匀,按拟订色谱条件进样测定。以对照品质量浓度(X,μg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程Y=20.825X+629.5,r=0.999 9(n=5)。结果表明,黄芩苷质量浓度在10.36 ~82.88 μg/mL 范围内与峰面积线性关系良好。

图2 黄芩苷高效液相色谱图
精密度试验:精密吸取上述对照品溶液,按拟订色谱条件连续进样6 次测定。结果黄芩苷色谱峰面积的RSD为1.10%(n=6),表明仪器精密度良好。
稳定性试验:取同一批(批号为20180319)样品,依法制备供试品溶液,按拟订色谱条件,分别于0,4,8,10,12,24 h 时进样测定。结果黄芩苷峰面积的RSD为1.90%(n=6),表明供试品溶液在24 h 内稳定。
从词性看,《北语词表》100词的名词与动词为主,合计占81%;而《鲁迅小说》词表100词中,如表2所示,名、动、代、副、连五类合计占81%。若按照一般语法分类,把时间、方位词和处所词归为名词,那么,《北语词表》100词主要为名词、动词和代词3类,而《鲁迅小说》词表主要是名词、动词、代词、副词和连词5类。
重复性试验:取同一批(批号为20180319)样品6 份,依法制备供试品溶液,按拟订色谱条件进样测定。结果黄芩苷含量的RSD为0.90%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的同一批(批号为20180319)样品6 份,各0.5 g,精密称定,分别加入对照品贮备液5 mL,再精密加入70%乙醇45 mL。按拟订方法制备供试品溶液,按拟订色谱条件进样测定,计算回收率。结果见表1。
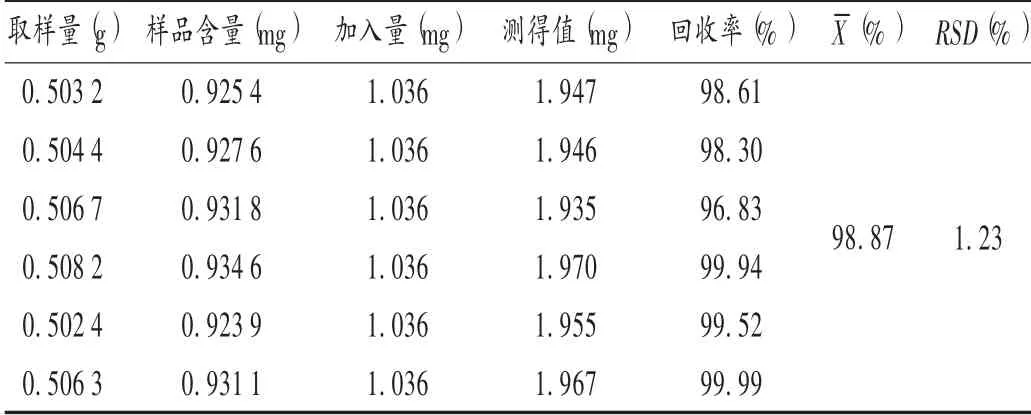
表1 黄芩苷加样回收试验结果(n=6)
2.2.4 样品含量测定
取 3 批(批号分别为 20180319,20180110,20180523)样品,依法制备供试品溶液,按拟订色谱条件进样测定,计算黄芩苷含量。结果3 批样品中黄芩苷含量分别为1.839,1.904,1.780 mg/g。
2.3 大黄素和大黄酚含量测定
2.3.1 色谱条件
色谱柱:Phenomenex Luna C18柱(250 mm×4.6mm,5 μm);流动相:甲醇(C)-0.1%磷酸溶液(D),梯度洗脱(0 ~20 min 时60%C→40%D,20 ~35 min 时60%C→40%D,35 ~55 min 时80%C→20%D);流速:1.0 mL/min;柱温:30 ℃;检测波长:254 nm;进样量:10 μL。
取大黄素对照品16.75 mg,精密称定,大黄酚对照品16.77 mg,置同一容量瓶中,加甲醇溶解制成质量浓度分别为33.50,33.54 μg/mL 的混合对照品贮备液;分别精密吸取混合对照品贮备液适量,加甲醇稀释成质量浓度分别为8.375,8.385 μg/mL 的混合对照品溶液。取样品适量,研细,取约10 g,精密称定,置具塞锥形瓶中,加8%盐酸溶液30 mL,超声处理2 min,再加三氯甲烷30 mL,加热回流1 h,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,洗液并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取5 次,每次10 mL,合并三氯甲烷液,蒸干,残渣加甲醇使溶解,转移至20 mL 容量瓶中,加甲醇稀释至刻度,滤过,摇匀,即得供试品溶液。按处方比例和工艺制备不含大黄饮片的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。
2.3.3 方法学考察
专属性试验:取上述3 种溶液各10 μL,按拟订色谱条件进样测定,记录色谱图。结果见图3,阴性对照无干扰,表明方法专属性良好。
线性关系考察:精密吸取混合对照品贮备液1.0,2.0,4.0,6.0,8.0 mL,分别置10 mL 容量瓶中,用甲醇稀释至刻度,摇匀。按拟订色谱条件测定。以对照品质量浓度(X,μg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程Y大黄素=11.21X+20.49(r=0.999 9),Y大黄酚=11.27X+22.93(r=0.999 9)。结果表明,大黄素、大黄酚质量浓度分别在3.35 ~26.80 μg/mL和3.354 ~26.83 μg/mL 范围内与峰面积线性关系良好。
精密度试验:精密吸取混合对照品溶液,按拟订色谱条件测定连续进样6 次。结果大黄素峰面积的RSD为1.50%(n=6),大黄酚峰面积的RSD为1.70%(n=6),表明仪器精密度良好。
稳定性试验:取同一批(批号为20180319)样品,依法制备供试品溶液,按拟订色谱条件,分别于0,3,9,12,18,24 h 时进样测定。结果大黄素峰面积的RSD为1.60%(n=6),大黄酚峰面积的RSD为1.40%(n=6),表明供试品溶液在24 h 内稳定。重复性试验:取同一批(批号为20180319)样品6 份,依法制备供试品溶液,按拟订色谱条件进样测定。结果大黄素含量的RSD为1.60%(n=6),大黄酚含量的RSD为1.60%(n=6),表明方法重复性良好。加样回收试验:取已知含量的同一批(批号为20180319)样品6 份,每份约5 g,精密称定,再取大黄素、大黄酚对照品适量,精密称定,加甲醇溶解制成每1 mL含大黄素8.421 μg、大黄酚13.742 μg 的对照品混合溶液,精密吸取对照品混合溶液2 mL,依法制备供试品溶液,按拟订色谱条件进样测定,计算回收率。结果见表2。
2.3.4 样品含量测定取 3 批(批号分别为 20180319,20180110,20180523)样品,依法制备供试品溶液,按拟订色谱条件进样测定,记录峰面积,并计算大黄素和大黄酚含量。结果3 批样品中大黄素含量分别为0.0330,0.0440,0.0299 mg/g,大黄酚含量分别为0.0528,0.0386,0.0496 mg/g。

图3 大黄高效液相色谱图

表2 加样回收试验结果(n=6)
3 讨论
3.1 TLC 鉴别
黄芩:分别考察50%甲醇、75%甲醇和甲醇作为提取溶剂,采用加热回流、超声方法提取,提取时间分别为0.5,1.0,1.5 h,考察乙酸乙酯-甲酸-水(9 ∶1 ∶1,V/V/ V)、乙酸乙酯-丁酮-甲酸-水(5 ∶3 ∶1 ∶1,V/ V/V/ V)、乙酸乙酯-丁酮-醋酸-水(10 ∶7 ∶5 ∶3,V/V/ V/ V)的上层溶液3 种展开剂。结果以甲醇为溶剂,超声处理30 min,乙酸乙酯-甲酸-水(9 ∶1 ∶1,V/ V/V)为展开剂所得溶液的TLC 图斑点清晰,分离度好,阴性对照无干扰。
冰片:分别采用正己烷、石油醚(30 ~60 ℃)作为提取溶剂,采用振摇、超声方法提取,提取时间分别为5,10,30 min。结果以石油醚(30 ~60 ℃)超声10 min 提取得到的TLC 图斑点清晰,分离度好,结果稳定。
3.2 含量测定
黄芩苷:分别考察50%乙醇、70%乙醇、乙醇作为提取溶剂,采用加热回流和超声方法提取,提取时间分别为0.5,1.0,1.5 h,考察甲醇-0.1%磷酸溶液(47 ∶53,V/ V)、甲醇-用磷酸调节pH 至2.7 的0.05 mol/L磷酸二氢钠溶液(42 ∶58,V/ V)[10]和乙腈-用三乙胺调节pH 至5.0 的0.02 mol/L 磷酸二氢钾溶液(14 ∶86,V/ V)[11]3 种流动相。结果以70%乙醇超声处理1 h,甲醇-0.1%磷酸溶液(47 ∶53,V/ V)为流动相得到的色谱图峰形对称,分离度大于1.5。
大黄素和大黄酚:分别考察甲醇-0.1%磷酸溶液和0.05%的磷酸水溶液-乙腈[12]2 种流动相,选用等度和梯度2 种洗脱方式。结果以甲醇-0.1%磷酸为流动相的梯度洗脱,大黄素和大黄酚理论板数高,峰形对称,分离度大于1.5。
