β2肾上腺素能受体的研究进展及其在兴奋剂残留检测中的应用
2020-03-13高海娇程古月袁宗辉华中农业大学国家兽药残留基准实验室HZAU农业部兽药残留检测重点实验室湖北武汉430070
高海娇,徐 超,程古月,袁宗辉 (华中农业大学 国家兽药残留基准实验室(HZAU)/农业部兽药残留检测重点实验室,湖北 武汉430070)
G 蛋白偶联受体(G-protein-coupled receptros,GPCRs)是一种嵌入到脂质双层结构且完整的膜蛋白,分布于组织最广泛的超大家族蛋白。根据跨膜结构的相似程度可分为以下5个亚家族:A 族视紫红样受体家族(包括α和β亚家族),B 族分泌素样受体家族,C族谷氨酸盐样受体家族,D 族黏附样受体家族和E 族frizzled/Taste2家族[1-2]。这些受体能够被胞外配体激活,包括蛋白,多肽,光,激素,离子和小分子等,从而通过细胞膜向G 蛋白传达信号[3]。β肾上腺素能受体(β-adrenergic receptor,β-AR)属于G 蛋白偶联受体A 族视紫红样受体,由7次跨膜螺旋组成。β-AR 具有各自的药理学、生物化学和分子生物学的特点,主要分为以下3 类:β1-AR、β2-AR 和β3-AR[4]。β1-AR 大 多 存 在 于 心 血管,主要引起血管舒张和增加心输出量,β2-AR 主要存在于支气管平滑肌以及骨骼肌,造成支气管舒张[5]。目前报道β3-AR 存在脂肪细胞,在调节并释放瘦蛋白发挥重要作用[6-7]。
肾上腺素受体兴奋剂与β-AR 有较好的亲和性,因此该受体广泛用于β肾上腺素类药物与蛋白的结合研究。本文将重点介绍β2-AR 的结构和功能,并总结该受体的表达纯化方式。分析β兴奋剂受体蛋白中部分氨基酸突变对蛋白活性的影响以及β2-AR 在兴奋剂残留检测中的应用。
1 β2肾上腺素能受体的结构及活性中心
1.1 结构组成β-AR 的组成包括:7 次螺旋跨膜(transmembrane,TM)结构,3个胞外环(extra-cellloop,ECL)和3 个 胞 内 环(inner-cell-loop,ICL)。β2-AR 含 有3 个 糖 基 化 位 点(Asn6、Asn15 和Asn187)以及2个额外的螺旋肽,即螺旋Ⅷ和ECL2中段的短螺旋片段[8]。蛋白的7次跨膜结构经过一系列的螺旋折叠,这些跨膜螺旋共同参与并形成一个“口袋”状配基结合位点。与药物的结合部位主要位于靠近胞外环的跨膜Ⅲ,Ⅴ,Ⅵ,Ⅶ以及胞外环ECL2[9]。靠近胞内的跨膜氨基酸及胞内环主要结合G 蛋白并参与G 蛋白信号的传导以及β-arrestin的招募[10]。β2-AR 以单体或者组成型二聚体形式动态平衡的存在于细胞膜上,与细胞膜上的蛋白结合位点相互作用,并且维持一定的表达水平[11]。部分GPCR 以二聚体才能发挥功能如C 族γ氨基丁酸受体B,但β2-AR 的单体形式能够保证蛋白发挥功能,并且与配体按1∶1结合[12-13]。
目前,人类的β2-AR 和鸟类的β1-AR 的晶体结构已经解析。以人源β2-AR 为例,跨膜Ⅰ至Ⅶ对应的氨基酸包括以下:TMⅠ29~60,TMⅡ67~96,TMⅢ103~136,TMⅣ147~171,TMⅤ197~229,TMⅥ267~298 和TM Ⅶ305~328,胞内环包括ICL1,61~66;ICL2,137~146;ICL3,230~266;胞外环包括ECL1,97~102;ECL2,172~196;ECL3,299~304;以及短肽螺旋Ⅷ[14]。ECL1,ECL2 和ECL3分别连接TM2、TM3、TM4、TM5 和TM5、TM6[15-16]。TM1的Lys60和螺旋8的Glu338氨基酸残基参与蛋白二聚体的形成,该位点的突变同N端羰基化位点突变产生相同的结果,进一步说明了N 端羰基化位点通过调节蛋白的二聚而影响蛋白功能[17]。在TM3和TM6胞质端,由(天冬氨酸/谷氨酸)精氨酸(色氨酸/酪氨酸)以及谷氨酸组成蛋白内部非共价键来稳定跨膜结构,β2-AR 是非保守序列,配基与蛋白结合时,离子锁被破坏,因此突变该位点时将改变蛋白活性[18]。
1.2 活性位点KOOISTRA 等[19]研究了GPCR晶体结构并预测蛋白配体的功能。通过晶体结构与同源模型的建立,证明了蛋白构象与配基结合的微小差异足够影响功能发挥。在结构模拟中,包含选定结合位点的氨基酸残基灵活性或使用多晶体结构或与不同配基结合的蛋白模型,从而系统得考虑了蛋白质构象和对接模拟的灵活性。目前人源β2-AR与配基结合的活性氨基酸以及作用机制已经通过分子模拟或者对接等手段研究清楚[20]。活性氨基酸主要位于TM3,TM4,TM5,TM6,TM7 以及ECL2,表1总结了β2-AR 存在的化学键以及与药物结合的活性氨基酸和作用方式。
1.2.1 二硫键 位于胞外环ECL2的2对半胱氨酸残基Cys106/Cys191 和Cys184/Cys190 形 成 二 硫键,分别与ECL1和TM3相互作用,稳定ECL2的结构[14]。早在1990年HENRIK 通过放射性配基结合试验与二硫苏糖醇竞争性结合β2-AR 受体,证明了有至少2对二硫键即Cys106-191,184-190参与了药物与配体的结合[21]。
1.2.2 氢键 β2-AR 与配基结合的主要作用力,包括Asp、Ser、Trp、Phe和Asn。PLAZINSKA 等[22]模拟了人源β2-AR 蛋白活化与非活化状态下与药物分子(非诺特罗)的结合模式,中心结合结构域包括TM3,TM5,TM6 和TM7。活化的Asp-113 既可以与带羟基的配基以氢键结合,Asn312也可以与羟基或者氨基以氢键结合,但在非活化状态下无法与药物相互作用[23]。Ser203,Ser204,Ser207 通过氢键键合了大部分的β兴奋剂药物,同时,Ser165与Ser207联合组成的活塞口袋也在蛋白发挥活性的过程中有着重要作用[24]。Trp286 和Phe290 残基以高度保守的Trp286 为中心形成芳香环结合位点,Trp286作为蛋白的活性拨动开关,其旋转异构体在蛋白活性与非活性状态中相应得发生变化,从而促使药物与蛋白结合[25]。
1.2.3 疏水作用力 疏水活性中心主要由Val114,Val117,Ile169 和Phe290 组 成。Val114 是 高 度 保守序列,与药物的乙醇胺单链或芳香环结合,该位点突变会降低与药物的特异性结合但不影响蛋白的表达或者折叠[26]。Val117,Ile169 和Phe290 分别与药物的氨基和苯环相互作用,形成疏水中心[27]。
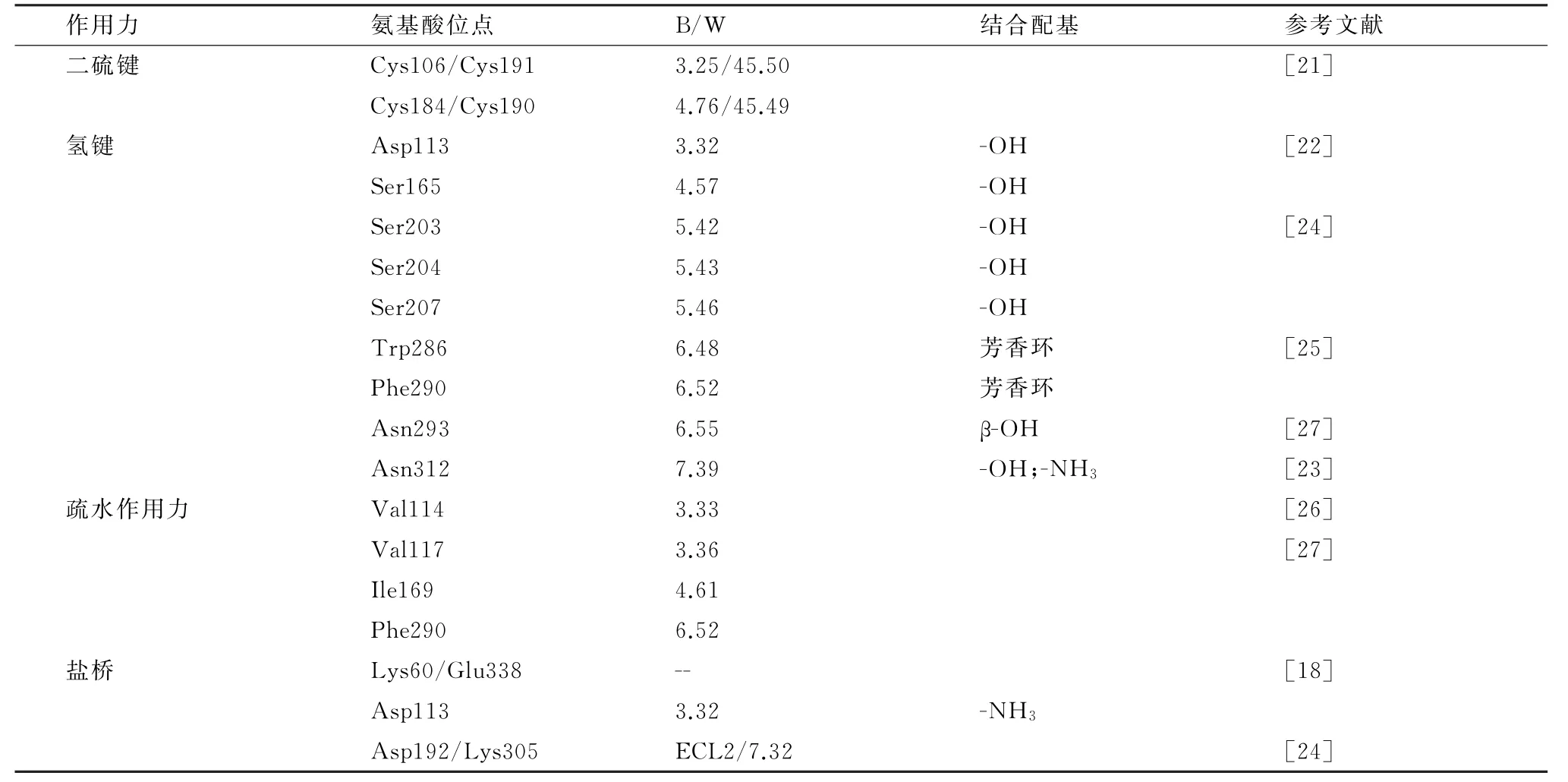
表1 人源性β2-AR 的活性氨基酸及其作用方式
1.2.4 盐桥 β2-AR 中参与盐桥形成的位点包括以下:Lys60(TM1)和Glu338(helix8),Asp113(TM3)以及Asp192(ECL2)和Lys305(TM7)。上述已经提及TM1的Lys60和螺旋8的Glu338参与了蛋白二聚体的形成,该氨基酸残基同时也在蛋白的一系列正确折叠后形成了盐桥。分子模拟中可观察到激动剂结合并激活蛋白的过程主要涉及2步,首先是配基上质子化的氨基与Asp113(TM3)相互作用形成盐桥,随其螺旋5旋转并暴露氢键结合位点与药物相连[24]。分子模拟配基通过蛋白内部的结合通道,发现ECL2中的Asp192和TM7的Lys305形成的盐桥正处于配基进入蛋白活性中心的通路中,当配基正准备结合蛋白时,则破坏该盐桥的形成,加速蛋白与配基的相互作用。不同类型的β肾上腺素受体模拟与药物结合的氨基酸序列有一定差异,总的仍存在于ECL2和TM7上的氨基酸[28]。
2 β2兴奋剂受体的表达方法
β2-AR 是7次跨膜蛋白,因其结构的特异性以及多次跨膜,该蛋白一直处于低水平表达。以下总结了目前β2-AR 的表达方法,主要分为真核、原核细胞表达,以及无细胞系统表达(表2)。
2.1 真核细胞表达
2.1.1 CHO 细胞系 Chinese hamster ovary cell line(CHO)为中国仓鼠卵巢细胞系稳定表达株。HOFFMANN 等[29]在CHO 细胞中稳定表达蛋白β1-AR、β2-AR 和β3-AR,表 达 量 分 别 为(0.37±0.08),(0.28±0.02)和(0.38± 0.08)nmol/g。BAKER 等[30]在前人的基础上,对火鸡β1-AR 进行突变,包括首尾氨基酸和内环ICL3 部分碱基的删除,以及热稳定性的6个碱基突变。在最终的突变体中,N 端删除30个氨基酸,6个热稳定性突变体得到的蛋白含量最高,达到2.79 nmol/g,而野生型蛋白仅达到0.15 nmol/g。说明对蛋白的氨基酸适当的进行增减以及突变,能够改变蛋白的表达量,但是与药物的特异性结合还需要进行后续分析。
2.1.2 HEK293 细 胞 Human embryonic kidney cells(HEK293)细胞为人源胚胎肾细胞。CHELIKANI等[31]构建了四环素诱导的HEK293高度表达仓鼠源β2-AR 蛋白的体系,该蛋白在膜蛋白中可得到蛋白(220.00±40.00)nmol/g,最终纯化后可得到活性蛋白12.40 nmol/g。另外,王迪[32]用四板细胞瓶(15 cm)培养HEK293 细胞并表达仓鼠源β2-AR 蛋白,最终得到纯化蛋白2.25 nmol/g,一次纯化 最 终 得 到 蛋 白135 μg。WANG 等[33]也 在HEK293细胞中表达猪源β2-AR 蛋白,但在膜蛋白中含有蛋白约17.00~23.00 nmol/g,一次得到纯化蛋白160.00μg。PARMAR 等[34]在β2-AR 的N 端添加了黄色荧光蛋白(YGP),在HEK293细胞中表达野生型与突变型的人源β2-AR 蛋白,野生型蛋白可得到(2.78±0.30)nmol/g蛋白,突变型蛋白的表达量比野生型降低了85%。
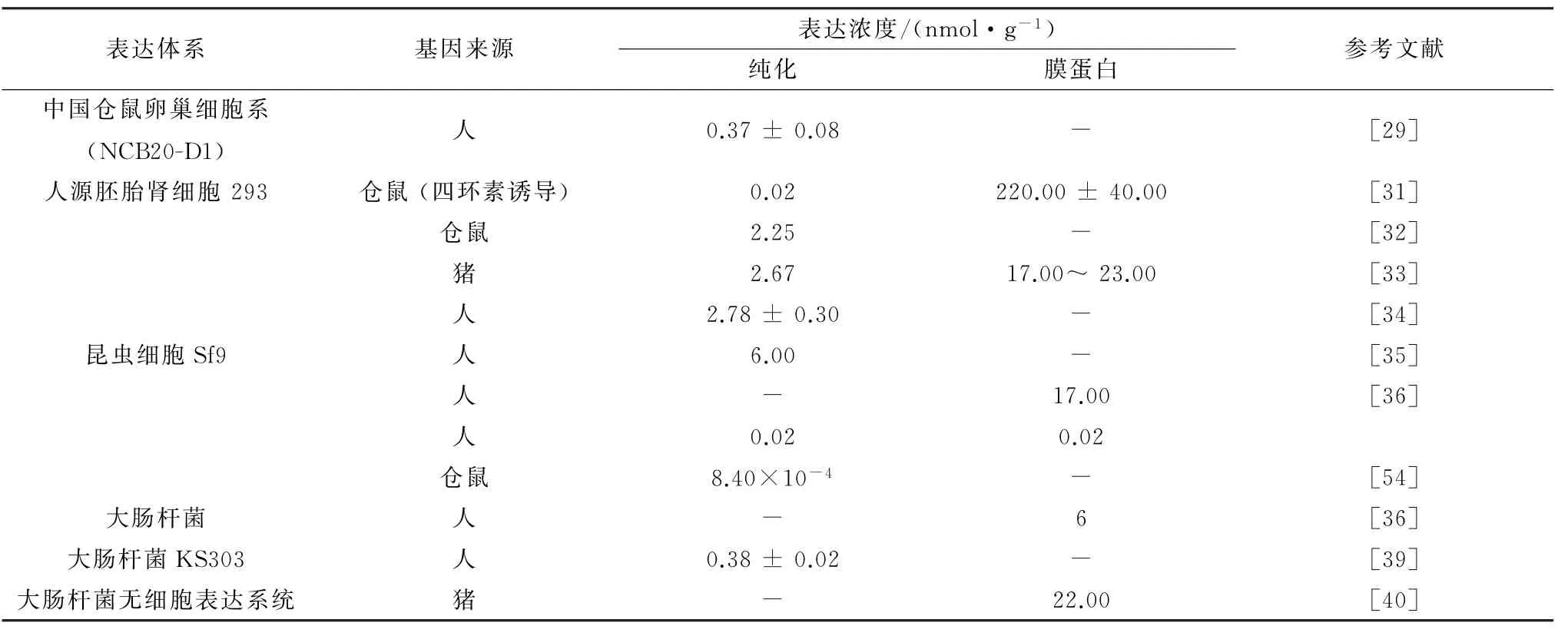
表2 β2-AR 的表达水平
2.1.3 Sf9细胞 杆状病毒-昆虫细胞系统是利用杆状病毒感染昆虫细胞(spodoptera frugiperda,Sf9)从而获得受体。该系统是目前表达G 蛋白较为成熟的一种表达体系,能够保证蛋白功能的完整性。KOBILKA 等[35]最初通过N 端添加16个短肽作为信号序列和Flag标签,C 端添加His标签,在昆虫-杆状病毒表达系统表达蛋白获得可溶性蛋白23.00 nmol,经过镍亲和纯化可获得具有亲和性的蛋白6.00 nmol。HAMPE 等[36]通过N 端麦芽糖标签,C端6His标签构建的β2-AR 融合蛋白分别在大肠杆菌和杆状病毒感染昆虫细胞中表达。杆状病毒获得了较大肠杆菌更高的表达量,大肠杆菌表达6.00 nmol/g,而 昆 虫 细 胞 则 表 达17.00 nmol/g。PARKER 等[37]在昆虫细胞中首次表达了一系列的C端截短的火鸡β1-AR 致使表达量有极大的提高,方便了去垢剂的溶解并且依然保持其调节活性,通过用亮氨酸代替116位非保守的半胱氨酸也会使表达量提高。为增加蛋白的表达量,可对氨基酸进行突变,通常是增加GC含量,或者在不影响蛋白高级结构,将疏水氨基酸突变为亲水性氨基酸。
2.2 原核细胞表达利用大肠杆菌表达真核β-AR蛋白比真核细胞表达简单快速,但是原核细胞无法正确折叠蛋白也不能修饰,无法保证蛋白功能的完整性。β2-AR 是7次跨膜蛋白,对于大肠杆菌来说,表达跨膜蛋白会对大肠杆菌产生一定的毒性,细菌会停止生长从而导致表达量低,蛋白错误折叠[38]。DANYI等[39]在基因改造后的大肠杆菌KS303 的内膜上表达重组人源β2-AR,并通过β兴奋剂与放射性配基竞争结合蛋白,最终可得蛋白浓度达到(0.38±0.02)nmo L/g。WANG 等[40]利用了大肠杆菌无细胞表达体系表达,通过优化猪源β2-AR 密码子使之尽可能多在大肠杆菌提取物中表达,表达量可达到1.10 g/L(约22.00 nmo L/g),但是蛋白的活性比真核细胞表达的蛋白低。大肠杆菌无细胞表达体系是模拟原核细胞的胞内环境,人为的不断添加底物和能量以维持环境的稳定,但因为缺少翻译后修饰导致蛋白活性不高。
3 β2兴奋剂受体的突变及亲和性研究
目前突变研究最多的是人源β2-AR,包括蛋白的跨膜区点突变和N 端截短突变,表3总结了β2-AR 受体蛋白相关突变研究,蛋白的亲和性主要参考抑制常数和半数有效量。
李晓娜[41]通过突变Asn6Gln和Asn15Gln除去N 端的2个糖基化位点,在HEK293细胞中表达后显示膜蛋白的表达水平无显著性变化(突变体蛋白的表达量为野生型的0.8倍),但降低了蛋白二聚体的形成,影响异丙肾上腺素作用下β2-AR 对G 蛋白信号的激活,而187位的糖基化位点不影响受体的二聚。YAO 等[42]对跨膜TM3和TM6区域组成离子锁的氨基酸进行突变,包括Ile135Trp、Ala271Cys,同时突变5 个位点的丝氨酸。在SF9细胞中表达的突变体破坏了离子锁,对于部分激动剂如沙丁胺醇来说不如完全激动剂如异丙肾上腺素的亲和性,异丙肾上腺素的抑制常数比沙丁胺醇小20多倍。TM2的122位谷氨酸是位于螺旋表面的非保守氨基酸,与TM 4-3-5螺旋上的氨基相互作用形成,维持空间结构。ROTH 等[43]研究该氨基酸突变为疏水氨基酸如色氨酸时,能近10倍得增加蛋白的热稳定性(野生型半衰期3.00 min,突变型Glu122Trp半衰期27.80 min),蛋白表达量也增加了1.5倍,但是药物的亲和性下降了50%,显示对异丙肾上腺素的抑制常数增大了1.0 倍。PARMAR等[34]突变TM1带电荷氨基酸和螺旋8的远测部,通过生物发光共振能量转移等方法测定蛋白二聚体的形成,突变Lys60Glu 和Glu338Ala后破坏了蛋白二聚体形成,使蛋白滞留在内质网。在众多的突变体中,蛋白的表达量与野生型蛋白相比相差不多甚至更少,蛋白的亲和力也没有得到很大的提升,表明该受体非活性氨基酸的突变对蛋白的表达量和亲和力没有明显改变。
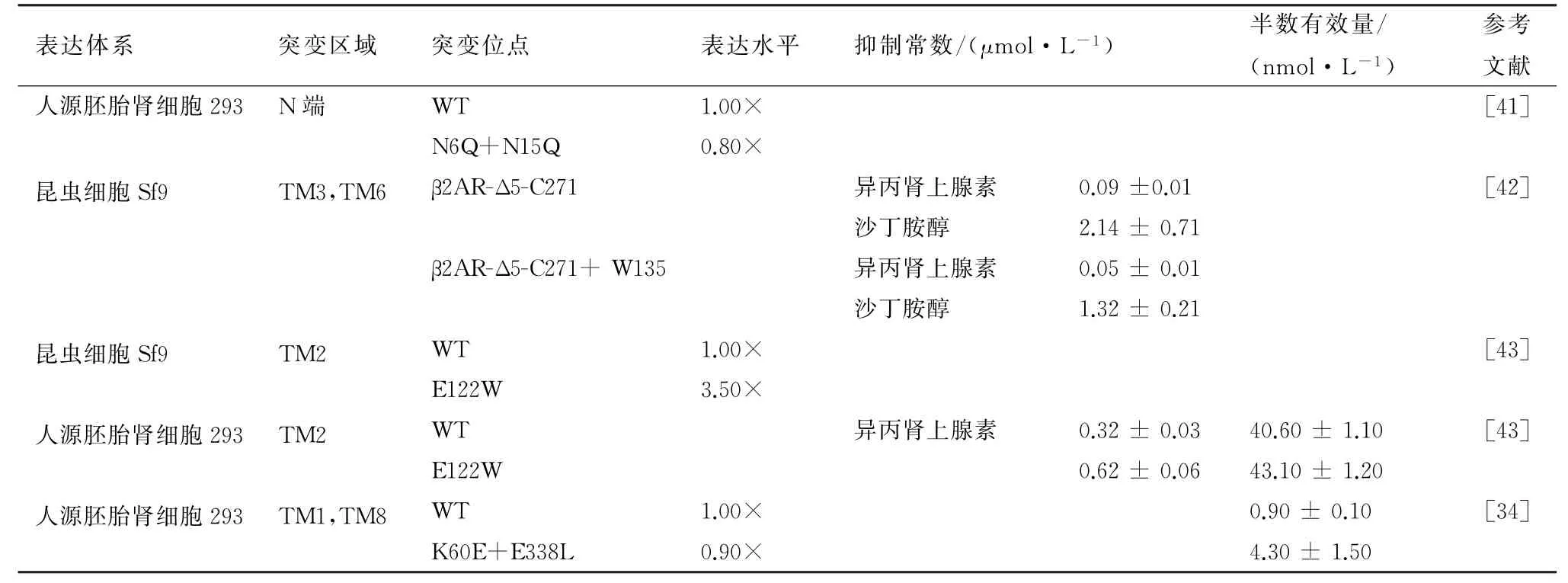
表3 人源性β2-AR 的突变研究
4 β2兴奋剂残留筛选的受体分析方法
目前,兴奋剂药物残留检测主要还是以仪器方法和免疫学方法为主。仪器方法包括高效液相色谱法(HPLC)和气/质联用分析法(GC/MS),最低检测限分别可达到盐酸克伦特罗0.25μg/L和莱克多巴胺0.50μg/L[44-45]。免疫分析法是以抗原抗体的特异性、可逆性结合为核心反应的分析方法,该方法具有快速简便、灵敏度高、特异性强、处理量大等特点。由于抗体的特异性高,只能同时检测一种或少数几种药物[46-47],如LIU 等[48]合成半抗原SAL 并获得抗体后建立酶联免疫分析方法,测得的沙丁胺醇的最低检测限为0.02μg/L。受体分析法是基于兴奋剂与β2-AR 结合,类似于免疫分析法中抗原与抗体的结合,一般采用β2-AR 作为受体,结合多种标记技术以提高灵敏度,如同位素标记和酶标记等[49](表4)。
4.1 放射性受体分析方法CHARM 法采用放射免疫受体分析技术检测兴奋剂的残留,通常先将待测样品与一定量的受体反应,然后加入一定量的同位素(3H 或125I)标记药物,反应平衡后,离心除去未与受体结合的标记药物,测定结合的标记药物的放射性,根据结合率推算样品中待测药物的含量[50]。二氢阿普洛尔(dihydroalprenolol,DHA)和氰基吲哚洛尔(cyanopindolol,CYP)作为β受体拮抗剂,能够特异性结合β2-AR,并能与其余的兴奋剂药物产生竞争作用。NODA 等[51]最早测定β2-AR 蛋白活性并获得高亲和性的野生型蛋白,与[125I]CYP 的亲和性Kd值为35.60 pmol/L。因此,该类方法皆通过放射性标记这2种药物并测得β2-AR 对配体的亲和性。
MEENAGH 等[52]在哺乳动物细胞系(CHO)表达受体蛋白,利用[3H]DHA 检测了沙美特罗、马布特罗、克伦特罗、西马特罗和莱克多巴胺,其中对沙美特罗的亲和性最高,半数抑制浓度(50%inhibition concentration(IC50))为87.50μg/L,抑制常数Ki为54.58μg/L,对莱克多巴胺的亲和性最低(IC50为3 313.25μg/L,Ki为20 722.89μg/L)。BOYD 等[53]在哺乳动物细胞系(NCB20-D1 cells)中表达人源性可溶蛋白β2-兴奋剂受体,也通过3H标记DHA 检测上述5种药,该类药物对沙美特罗的亲和性最高,其IC50为112.90μg/kg,最低检测限为12.10μg/kg。DANYI等[39]在大肠杆菌KS303表达人源性可溶蛋白β2-兴奋剂受体,通过[125I]CYP检测该类药物,测得对克伦特罗的亲和性最高(IC50为15.67μg/L,Ki为6.58μg/L)。放射性受体分析方法灵敏度高,常用于检测兴奋剂与β2-AR 受体的竞争性结合,并研究后续G 蛋白偶联受体的信号传导功能[39]。但该方法采用了放射性同位素标记药物,容易造成放射性污染并对人体有一定的伤害,阻碍了实际生产应用。
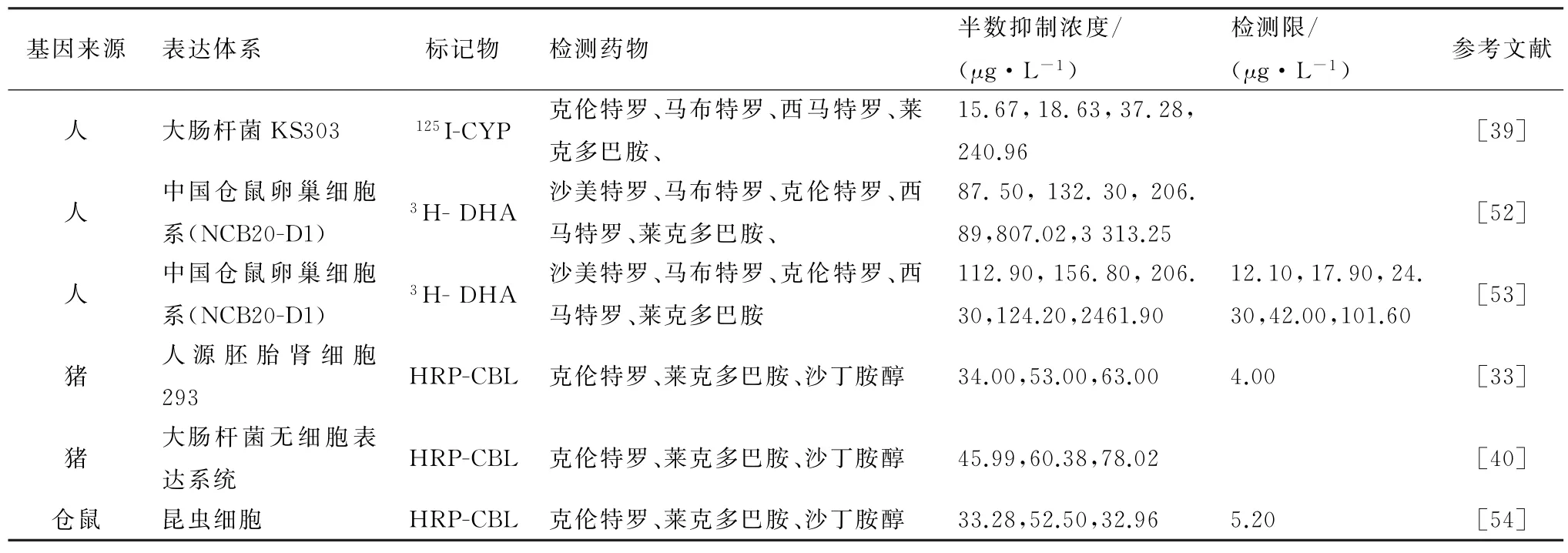
表4 β2-AR 的受体分析方法
4.2 酶标记受体分析方法酶标记受体法是基于受体配体间特异性相互作用和酶标记技术而建立的一种检测技术,类似于ELISA 检测方法。通过包被物即受体蛋白,酶标记物即常规的辣根过氧化物酶(horse radish peroxidase,HRP)以及竞争方法等方面的不同组合建立检测模式。
CHENG 等[54]采用仓鼠的β2-兴奋剂受体作为受体蛋白,在昆虫细胞SF9 中表达,表达量为50.00μg/L。分别以HRP-克伦特罗,HRP-沙丁胺醇,HRP-莱克多巴胺作为酶标记药物,建立了直接竞争方法,分别检测这3 类药物,孵育检测时间为1 h,IC50分别为33.28,52.50和32.96μg/L,其中对莱克多巴胺识别力最强,其最低检测量达到5.20μg/L。WANG 等[33]采用猪肾细胞的β2-兴奋剂受体作为受体蛋白,在人胚肾细胞(HEK293)中表达。以HRP-克伦特罗为酶标记药物,用于检测猪肝肾组织中的克伦特罗,沙丁胺醇和莱克多巴胺,IC50分别为34.00,53.00和63.00μg/L,最低检测量达到4.00μg/L。WANG 等[40]克隆猪肾细胞的β2-兴奋剂受体,采用无细胞表达蛋白系统纯化表达,纯化后的蛋白含量高达800.00 mg/L(约16 nmol/g),与真核细胞HEK239 细胞的总膜蛋白表达量(17.00~23.00 nmol/g)近似,但是无细胞表达后的蛋白活性比在HEK293中降低10%~20%,对克伦特罗,沙丁胺醇和莱克多巴胺的IC50分别为45.99,60.38和78.02μg/L,说明无细胞表达纯化的活性蛋白含量减少。
5 展望
β2-AR 受体是G 蛋白偶联受体最重要的一员,在受体信号转导中发挥重要作用。但是因为7次跨膜结构导致不能大量异源表达,阻碍了蛋白的研究发展。本文综述了β2-AR 蛋白在原核以及真核细胞的表达,真核细胞能够保证蛋白活性,但是蛋白表达量只能达到nmo L/g,远远达不到蛋白的需求。在蛋白的突变研究中,包括点突变和截短突变,表达量和亲和力与野生型蛋白相差不大,还需要深入研究。因该蛋白较强的生物活性和亲和力,能够与兴奋剂相互作用,因此广泛用于兴奋剂的残留筛选。目前检测的药物包括克伦特罗,莱克多巴胺,沙丁胺醇,最低检测限为4μg/L,但是对其他兴奋剂药物的识别还不能达到检测水平。放射性标记检测结果比酶标记更为精确,但放射性物质污染环境不能投入使用,只能在实验室检测中使用。因此,还需要继续开发检测方法,优化检测条件。
G 蛋白偶联受体在生物体内发挥重要的信号转导作用,因此需要更加清晰研究7次跨膜蛋白的分子结构以及与小分子药物之间的相互作用机理。同时开发或改造不同的细胞表达系统为扩大蛋白表达做准备。
