问答式审评体系在药品技术转让审评的应用探索
2020-03-11王晓英陈佩毅许庆锐王爽张一凡林焕冰
王晓英,陈佩毅,许庆锐,王爽,张一凡,林焕冰
(广东省药品监督管理局审评认证中心,广东 广州 510080)
为有效合理地利用审评资源,提高仿制药药学审评的质量和效率,美国食品药品监督管理局(FDA)仿制药办公室(The Office of Generic Drugs,OGD)针对简单剂型、复杂剂型、窄治疗窗的仿制药,在质量源于设计理论指导下,设计并提出了问答式审评质量评价体系(question-based review,QbR)。QbR遵循了FDA的药品生产质量管理规范(current Good Manufacture Practice,cGMP)[1]的基本概念和原则以及PAT(process analytical technology)的理念,用于仿制药的药学部分(chemistry,manufacturing and controls,CMC)的技术审评[2],审评过程中重点关注药品的关键质量属性。QbR不仅能提高对仿制药上市申请的审评质量及审评效率,还能引导仿制药生产企业沿着正确的方向开展相关的研究工作,最终保证产品质量和疗效,实现研究、评价及产品的多重效益。
为促进新药研发成果转化和生产技术合理流动,鼓励药品生产企业兼并重组,提高竞争力,激发市场活力,国家药品监督管理局分别于2009年颁布了《药品技术转让注册管理规定》[3]和2013年颁布了《关于做好实施新修订药品生产质量管理规范过程中药品技术转让有关事项的通知》[4]。近年来,随着药品供给侧改革的深入,药品生产技术转移数量呈现逐年上升趋势,而各省的药品审评机构的审评资源已出现相对不足现象。如何加快药品生产技术转移品种上市的审评及批准,同时确保药品生产技术转移前后品种的同质同效性,已成为各省药品审评机构所面临的亟待解决的实际问题,因此有必要通过借鉴和吸取先进的管理理念和管理手段,改革原有的审评模式,探索创建一个有的放矢的风险药品转移技术评价体系,有效合理地利用审评资源,提高审评的质量和效率。
QbR是FDA为提高仿制药上市申请的审评质量及审评效率,在质量源于设计理论指导下开发的新的审评模式。在FDA运用QbR在仿制药的审评实践获得成功后,国家药品监督管理局药品审评中心(CDE)也借鉴FDA开发的QbR审评模式,在质量源于设计理论指导下,针对糖盐水类注射液开发了新的药品技术评价体系,并获得预期的效果。因此,借鉴QbR的审评模式,结合药品技术转让审评的技术特点,探索QbR在药品技术转让审评的应用,对解决药品生产技术转让存在的实际问题具有重大意义,且对我国现有药品审评制度改革提供新思路。
1 QbR开发背景
21世纪初,FDA提倡cGMP的核心目标是为了保证良好的药品质量,并且更为重视实现这一目标的全过程。但FDA通过全面分析对比实施cGMP质量目标要求与原仿制药审评工作之间的差距,意识到原实行的仿制药审评工作模式无法满足实施cGMP质量目标要求。例如,在原审评模式下,评价一个药品的质量优劣,主要通过产品的终点检验来判定产品的质量和性能,很少关注如何设计一个有效和有效率的制造过程,以确保药品的稳定良好质量[5]。然而,在cGMP实施过程中,药品质量的概念是贯穿整个生产过程中的一种行为规范。因此,在原审评模式下一个质量完全合格的药品,也未必是符合cGMP质量要求。因为它的生产过程中可能存在偏差,其潜在的风险是不能被检验报告所发现的。原审评模式忽略了产品检验质量标准与产品的生产工艺关键质量属性相关性;同时也忽略了所申请产品的风险大小,减低审评资源的有效利用,即无论风险高低制剂均采用相同的方法和程序进行审评,且提出的要求往往带有一定的强制性,导致低风险产品浪费过多审评资源,高风险产品占用的审评资源不足[1]。
与此同时,21世纪初,FDA仿制药办公室的评审工作量呈逐年快速增长的趋势。OGD在2002年共收到361个ANDA。这个数字在2003年增加到449个,在2004年增加到563个[5]。而且随着获批仿制药品数量增加,需要再次递交变更申请的数量也随之增加,这将进一步增加了FDA对每个产品的审评循环与审查轮数,增加了审评工作负荷,浪费了审评资源。因此,FDA 对仿制药CMC审评模式进行改革,创建一个聚焦于药品质量关键属性的风险审评模式,即QbR。新的审评模式不仅为cGMP实施奠定了坚实的基础,并且有效配置有限的审评资源,提高审评的质量和效率。
2 QbR在药品技术转让审评的应用优势
QbR审评模式聚焦于药品质量的关键质量属性评审,其鼓励审评人员识别出保证产品质量所必须的关键标准和生产控制,并将对产品进行风险评估,设计并明确技术审评需要提出的重点问题及需要解决的问题,有效合理地利用审评资源[6]。QbR在药品技术转让审评的审评应用,应基于质量源于设计的理念,通过建立固定化的QbR问题和格式,指导审评员提出正确的问题,提高审评员的判断力,从而使他们能够更好地识别出转让双方在CMC信息中影响产品质量的缺陷;通过鼓励申请人分享其药品技术转让过程中的知识,如药品技术转移报告、技术转移文件等,促进理解处方和生产工艺的因素如何影响药品质量的机理,从而建立更具有相关性的质量标准和生产过程控制;通过纳入风险评估环节,将法规监管与科学认知水平和制剂的复杂度相关联,从而释放紧缺的资源。
3 QbR在药品技术转让审评的应用开发
OGD 在QbR的应用开发时遵循以下4个原则:①质量由设计、开发和生产构建而成,并通过测试确认;②基于风险评估方法可以最大限度地节省时间、精力和资源;③尽量保留原审评系统的最佳做法;④通过充分利用现有的科学和广泛的咨询,确保高质量的审评。因此,QbR在药品技术转让审评的应用开发过程,也应遵循QbR开发的4个基本原则。首先要识别现有药品技术转让的审评过程的核心问题,即如何保证受让方能持续稳定生产出与转让方质量一致的产品。以这一核心问题为基础,识别分析与此相关的审评各个关键方面。然后,对比分析审评核心问题与原审评体系存在的差距,分析原审评体系的最佳做法,揭示出未解决的关键科学问题。识别了关键问题后,采用基于风险的方法,确定哪些问题适合哪些产品。设计和起草QbR文件,经审核后,听取审评员和申请人的意见,在试点评估前进行广泛征求申请人和公众意见。经培训后,审评机构在部分品种中进行试点评估。最后经试点评估修正后,审评机构将全面实施。
4 QbR在药品技术转让审评的应用的问题提纲
目前在仿制药品上市申请的分类前提下,FDA 已经针对简单剂型、复杂剂型、窄治疗窗药品制定了“审评提问问题提纲”[7],并且编制了口服速释剂型[8]及缓释胶囊[9]的质量综述报告样板和最终灭菌产品的质量综述报告样板。
现本文通过借鉴和吸取FDA制定“审评提问问题提纲”中的优点,结合现有我国药品技术转让的法规和相关技术指导原则要求,制定药品技术转让审评提问问题提纲(详见表1和表2),有利于有效地利用有限的审评资源,使审评审批规范化、标准化,并能为申请人提供申报指引,提高申请人的申报水平。
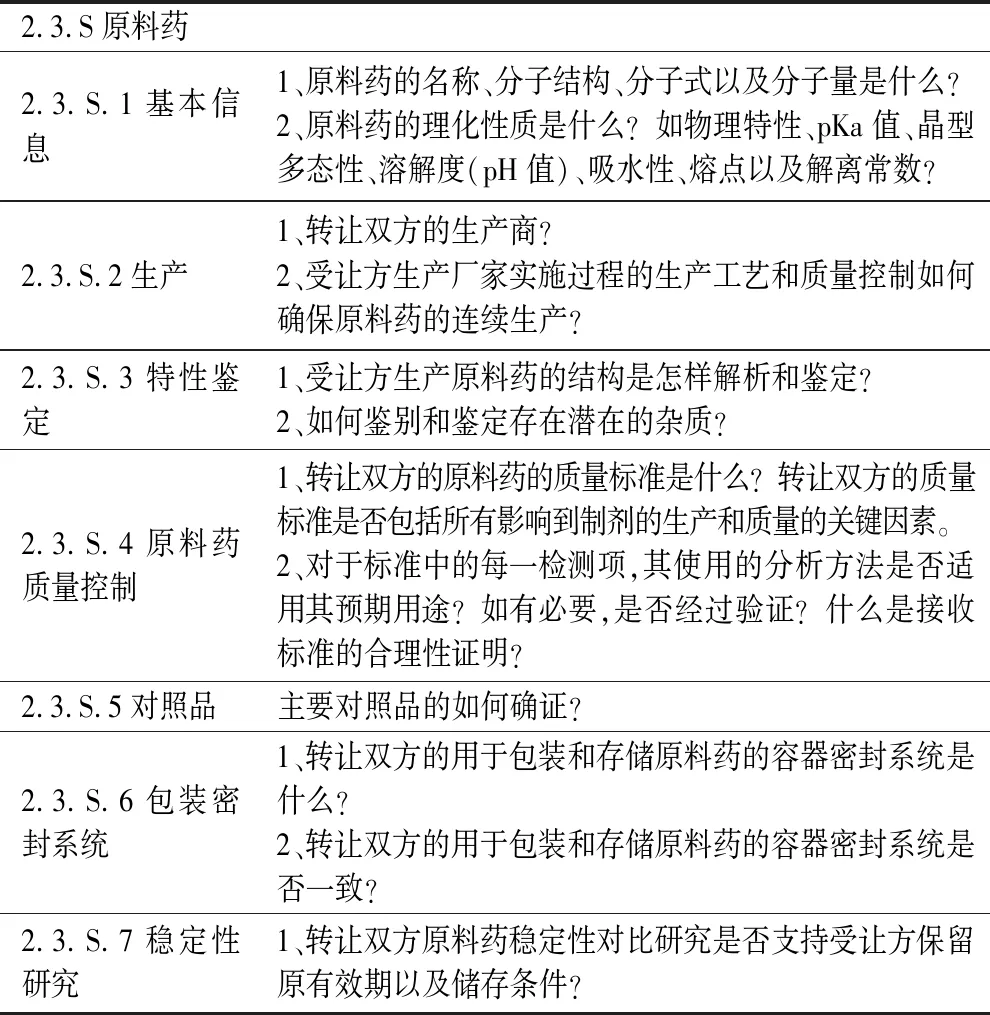
表1 原料药药品技术转让审评提问问题提纲
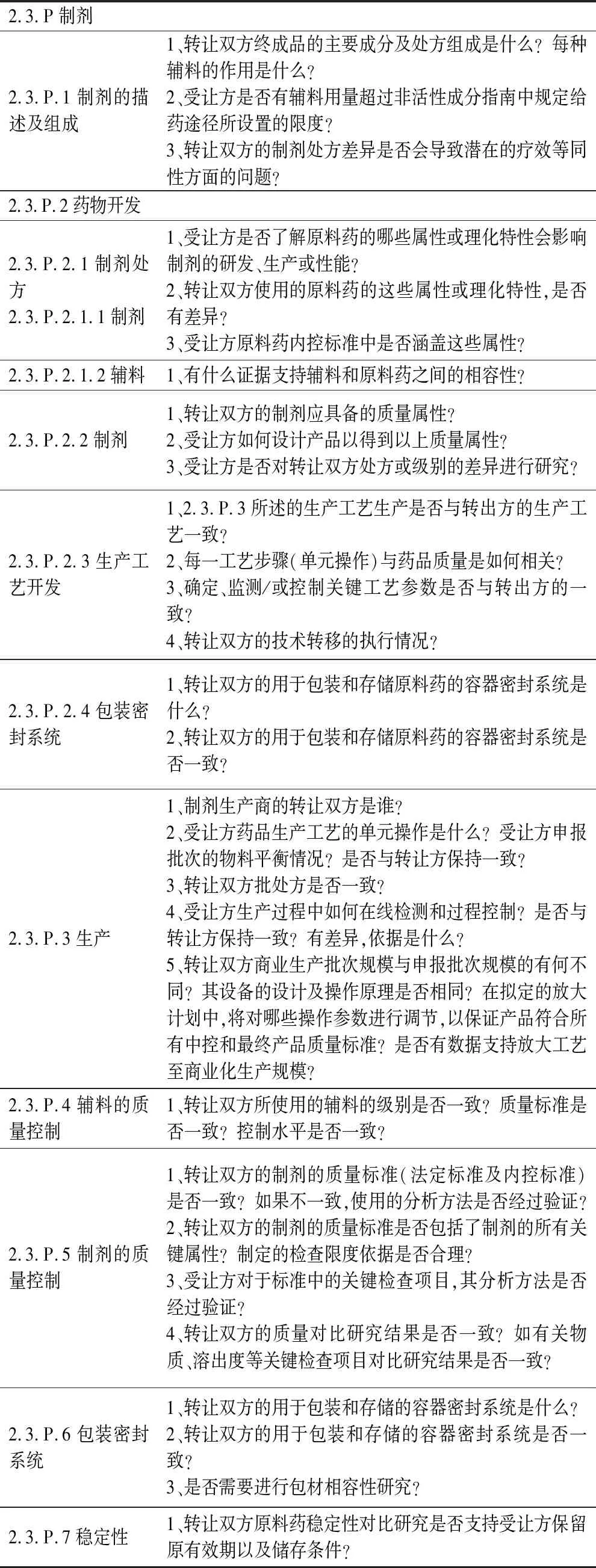
表2 制剂药品技术转让审评提问问题提纲
5 结语
当前,我国医药产业快速发展,创新创业方兴未艾,审评审批制度改革持续推进。但总体上看,我国医药产业科技创新支撑不够,上市产品质量与国际先进水平存在差距。为促进医药产业结构调整和技术创新,提高产业竞争力,满足公众临床需要,如何在充分了解美国等发达国家对药品审评管理思路与技术要求的基础上,探索出符合我国国情的药品审评管理思路,已成为当前重要的研究方向。
FDA提出的仿制药上市申请的QbR对我国在药品审评管理理念上的更新及在管理手段、方法上的改进和完善,具有较大的参考意义,通过借鉴和吸取其中的优点和长处,可以提高我国的药品监管质量与效率。
