卡托普利:从多肽到小分子降血压药物的研发历程
2020-03-11文雨刘健胡立宏
文雨,刘健,胡立宏
(南京中医药大学药学院中药功效物质重点实验室,江苏 南京 210023)
高血压是一种以动脉血压持续升高为主要表现的慢性疾病。目前临床上高血压主要分为两大类:原发性高血压和继发性高血压。其中原发性高血压占高血压患者90%以上,且发病机制尚不明确,可能与神经机制、肾脏机制、激素机制、血管机制、胰岛素机制等密切相关。患者由于血压升高,心脏需超负荷工作,以便泵送足够的血液及氧气来维持正常的身体机能。如果不及时治疗,可能会导致心脏相关疾病,并伴随肾脏损伤,危害大脑和眼睛等器官。据世界卫生组织报告,1980年全球有6亿人患有高血压,而2008年患病人数升高到10亿,预计到2020年这一人数可能会达到15亿[1]。在我国高血压的患病人数也一直居高不下,因此抗高血压药物的开发具有巨大的社会及经济价值。
人类对血管紧张素转化酶(angiotensin-converting enzyme,ACE)作用机制的解析被认为是高血压治疗的重要突破。这一发现主要是基于对体内肾脏中的肾素-血管紧张素-醛固酮(renin-angiotensin-aldosterone system,RAAS)系统的认识。RAAS是肾脏产生的一种升压调节体系,肾素通常是由肾脏内的肾小球旁器官产生和分泌,以应对血压下降或过滤后的氯化钠浓度下降。一方面,分泌的肾素将肝血管紧张素原裂解成无活性的血管紧张素Ⅰ,然后通过ACE将血管紧张素Ⅰ转化为活性的血管紧张素Ⅱ。血管紧张素Ⅱ有许多生理作用,其增加血管阻力,作用于肾上腺皮质释放醛固酮,刺激后垂体释放抗利尿激素加压素,刺激口渴中枢,促进交感神经末梢去甲肾上腺素释放,抑制去甲肾上腺素的再摄取。同时,血管紧张素Ⅱ也作为激动剂将其受体激活,从而引起一系列相应的生理变化,最终导致血管壁紧缩、血压升高。另一方面,血管缓激肽本身是具有促进血管扩张、降低血压的作用。而ACE可促进缓激肽的降解、失活,间接引起血压的升高[2-3](见图1)。
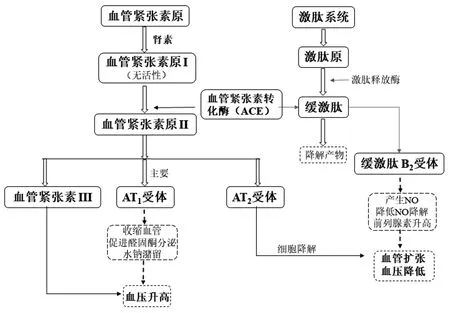
图1 肾素-血管紧张素-醛固酮(RAAS)系统对血压的调节机制
血管紧张素转化酶抑制剂(angiotensin-converting enzyme inhibitors,ACEI)可作为降压药物,其降压原理主要是通过阻断RAAS系统,抑制ACE 活性,从而抑制循环及局部组织血管紧张素Ⅰ转化为血管紧张素Ⅱ,使血管紧张素Ⅱ生成减少,抑制缓激肽降解,从而具有舒张血管、降低血压的作用[4]。卡托普利(商品名 Captopril)是第一代口服ACEI,其最初来源于巴西蛇毒中的一种缓激肽,能够有效地治疗各种原发性高血压,具有起效迅速、降压平稳、远期疗效突出等优点,同时对心力衰竭和糖尿病、肾病也有良好的治疗效果。卡托普利的问世开启了高血压药物治疗的新时代[5-6]。时至今日,以卡托普利为代表的ACEI的研究,仍是药物化学研究的热点之一。尽管ACEI、钙离子通道拮抗剂、血管紧张素Ⅱ受体拮抗剂等各类抗高血压新药层出不穷[7],但卡托普利仍在高血压的临床治疗中扮演着重要的角色。
1 卡托普利的研发历程
1.1 苗头化合物teprotide发现 早在1933年,Rochae Silva发现被巴西蝮蛇咬伤的患者会因为低血压休克而死亡,由此猜想蛇毒中应该含有一种“降压物质”。直到1948年,Rochae Silva终于成功地从巴西蝮蛇的蛇毒中提取出这种“降压物质”,并通过结构鉴定,确证该物质是一种直链的九肽化合物,命名为“缓激肽(bradykinin,简称BK)”。BK只有在蛇毒体内才能稳定存在,在人体内的半衰期极短,仅几分钟便会完全分解。因此,研究人员猜测毒蛇体内可能存在一种稳定BK的生源物质,于是对生源物质展开了研究。
1960年,Ng和Vzne发现巴西蛇毒中含有一种“缓激肽增强因子(bradykinin potentiating factor,简称BPF)”,同时,研究结果表明,BPF可在一定程度上抑制缓激肽降解酶,从而减缓缓激肽的降解[8-9],并且发现该化合物可与体外制备的血管紧张素转化酶反应,最终发现了有效的血管紧张素转化酶抑制剂。基于此研究结果,Ferreira等[10-12]先后从蛇毒提取液中分离得到不同的缓激肽增强因子,并且报道了这些长肽的氨基酸序列的合成。综合比较几种多肽后,发现五肽BPP5a对ACE抑制活性最好,但其体内的半衰期非常短。而九肽SQ20881(teprotide)抑制活性仅次于BPP5a,但却拥有相对较长的体内半衰期(T1/2=2.5 h)。
随后Vandongen和Greenwood[13]两位科学家开展了teprotide对肾灌注大鼠和异丙肾上腺素刺激肾素分泌的大鼠的影响。研究发现,减小teprotide的用量时,相关的大鼠血压升高,表明在肾中血管紧张素Ⅰ被转化为血管紧张素Ⅱ。另外,血管紧张素Ⅰ的转化也会影响肾素分泌和对异丙肾上腺素的刺激反应。最终随着teprotide临床试验的不断推进,研究人员确证teprotide是一种有效的ACEI,但因为它是一种大分子肽类化合物,存在肽类药物的缺点,如:透膜性差、生物利用度低、只能通过注射给药,从而限制了其在临床上的使用。
1.2 先导化合物1的确定 为了克服肽类药物的缺点,该类药物研发的重点开始转向可口服的小分子ACEI的开发,研究策略主要是探究teprotide的药效团,简化化合物结构,降低分子量,提高成药性。由于该多肽是大分子,可能具有一定程度的二级或三级结构,使其与ACE的活性位点之间进行特异的结合。生物学的研究发现血管紧张素与羧肽酶A均为肽类外切酶,这两种酶在序列上具有45.2%相似性,而当时羧肽酶A相较血管紧张素的研究较为充分,羧肽酶的蛋白结构已被成功解析,并且已有相关的羧肽酶A抑制剂报道。但由于当时计算机辅助药物设计技术并不成熟,研究人员也无法基于羧肽酶的结构去对血管紧张素进行同源模建,获得血管紧张素的蛋白结构,研究人员无法基于ACE的蛋白结构进行化合物设计。因此,Cushman和Ondetti只能先探讨teprotide与羧肽酶A活性位点可能的相互作用模式,用来指导teprotide的结构优化[14]。
羧肽酶A中正电性的Arg145与teprotide中C-末端负电性的苄基琥珀酸形成静电相互作用,同时羧肽酶A中疏水性氨基酸与底物中的苯环形成的疏水相互作用(见图2)。此外,羧肽酶A的Zn2+与底物的羰基相互作用,并促进该位点的酰胺键水解。尽管血管紧张素转换酶与羧肽酶A的活性位点非常类似,但其中也存在一定的差异[15]:①对于羧基静电相互作用区以及Zn2+结合区之间的距离,ACE大于羧肽酶A,因此在ACE中,Zn2+可能参与N-末端倒数第二个氨基酸水解,而非最后一个肽键的水解(见图3);②ACE及羧肽酶A中与底物疏水侧链结合的疏水口袋差别较大,ACE为圆形,羧肽酶A为六边形口袋(见图2a、2b);③在血管紧张素转化酶的活性位点上存在氢键供体与底物的羰基形成的氢键相互作用,羧肽酶A并无该氢键作用;以上的蛋白与小分子结合作用差异,证明ACE抑制剂不同于羧肽酶抑制剂,ACE抑制剂至少应具有二肽的结构单元。
Cushman和Ondetti综合分析苗头化合物teprotide和BPP5a的结构、BPF的构效关系以及受体与配体的结合模式,得到了关键酶结合底物Ⅰ(substrate Ⅰ)。Ⅰ有以下特征:①化合物C端的脯氨酸处于ACE的结合口袋,对整个化合物的活性至关重要,是关键的药效基团,负电性羧基不仅与靶蛋白中正电性Arg145残基形成静电相互作用,同时脯氨酸侧链还伸入疏水空腔中,与靶蛋白形成疏水作用;②与脯氨酸相连接的丙氨酸,则通过与靶蛋白形成氢键相互作用,而发挥抑制活性。但该底物对ACE的抑制活性较弱,IC50仅为600 μmol·L-1,因此该化合物仍需进一步活性优化。
Cushman和Ondetti进一步参考了羧肽酶A和其抑制剂的结合模式,并模拟ACE与底物Ⅰ的结合方式,经过生物电子等排设计得到先导化合物1,该化合物对ACE具有一定的抑制活性(IC50=330 μmol·L-1)。
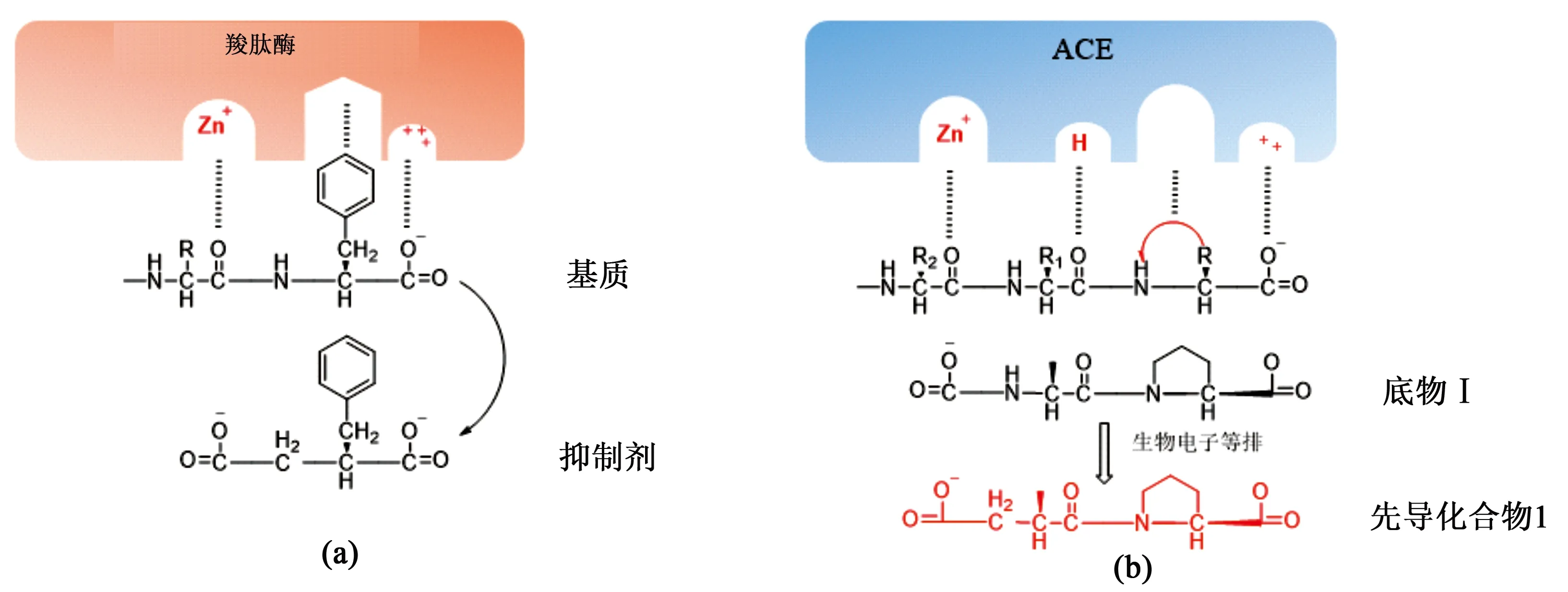
图2 a.羧肽酶A活性结合位点;b.血管紧张素转化酶活性结合位点
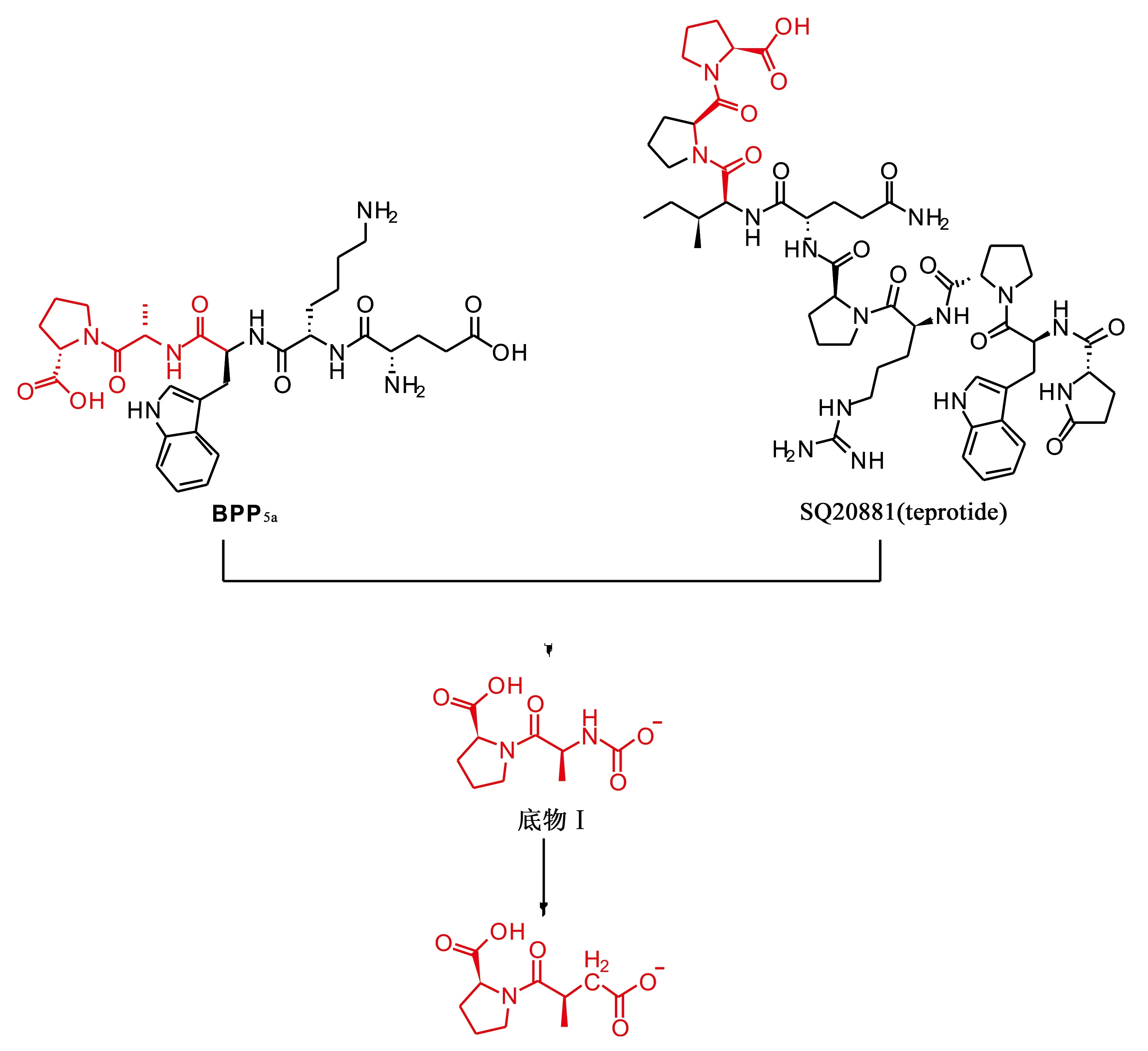
图3 脯氨酸药效团、底物Ⅰ、先导化合物1的发现
1.3 先导化合物1的结构优化 先导化合物1尽管活性仍较差,但从结构上已经较之前的九肽简化很多,以此为先导进行结构优化,有望发现新型的ACEI。通过前期对ACE的作用机制研究,发现ACE的活性中心会有5个结合位点。为了获得活性及理化性质更优的候选药物,Cushman和Ondetti对先导化合物1的5个结合位点分别进行了结构优化,设计、合成了一系列新的衍生物。
1.3.1 ACE正电活性位点和疏水空腔的结合基团优化 通过对该作用位点的结构分析,发现配体基团伸入ACE的结合口袋,一般是以氨基酸取代最优。因此,Cushman和Ondetti设计了一系列不同氨基酸取代的衍生物,期望能够获得活性更优的化合物(见表1)。
表1是不同氨基酸残基取代后的活性结果:从整体来看,疏水性越强的氨基酸进行取代,活性越好。结果显示:脯氨酸取代的活性最好,可能由于脯氨酸的五元杂环与疏水口袋更加疏水结合更强,将脯氨酸替换为其他氨基酸残基反而会降低化合物活性;另外比较L构型和D构型的脯氨酸和苯丙氨酸,发现L构型抑制活性远优于D构型,表明L构型氨基酸更易与ACE正电活性位点,形成更好的空间匹配与能量契合。

表1 不同氨基酸残基取代对ACE的抑制活性数据
注:*使用的是兔血管紧张素转化酶
1.3.2 依据与活性中心的Zn+的螯合作用进行结构优化 ACE的活性位点结构和羧肽酶A较为类似,依据苄基琥珀酸与羧肽酶A的结合特点,苄基琥珀酸会与活性中心Zn2+进行螯合,而该配位键作用相比氢键作用、疏水作用以及范德华作用更强,因此该配位键作用较为重要。因此,与ACE活性中心Zn2+形成配位的基团对整个化合物的活性至关重要。于是,Cushman和Ondett对该作用位点的基团进行一系列的替换,主要是引入Zn2+形成配位键的基团,如:-COOH、-SH、磷酸基团等,相关化合物结构和活性对应关系如表2所示。
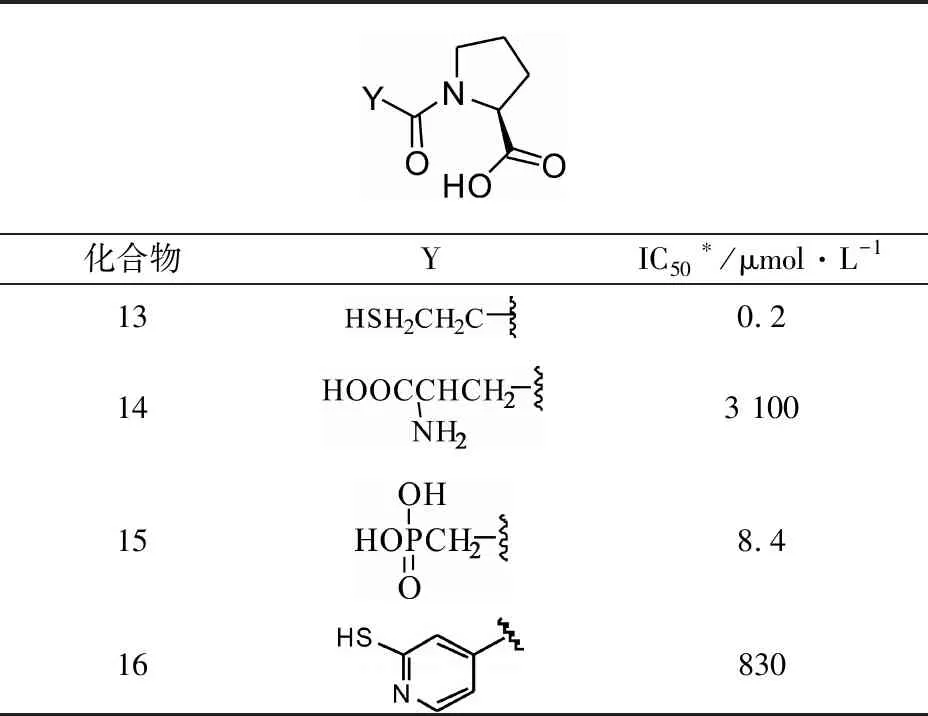
表2 不同基团和Zn离子结合的活性数据
注:*使用的是兔血管紧张素转化酶
活性测试结果表明,该部分的取代基对化合物活性影响较大。磷酸基团取代的化合物15较先导化合物1,体外酶活性提升了40倍左右;而巯基取代的化合物13比先导化合物1的体外酶活性提高了约1 600倍左右。表明巯基和活性位点的Zn离子结合最好,磷酸次之,羧酸和Zn离子结合效果最差,后期的结构优化主要以巯基为主。
1.3.3 脂肪链长度对活性影响 考虑到脂肪链的长度一方面直接影响整个化合物的理化性质,另一方面影响巯基与Zn2+的相互作用以及C-末端脯氨酸与Arg的静电相互作用、疏水相互作用。因此,研究人员对该位置的脂肪链长度进行延长或缩短,期望能够进一步增强化合物与相互作用,从而提高化合物活性(见表3)。
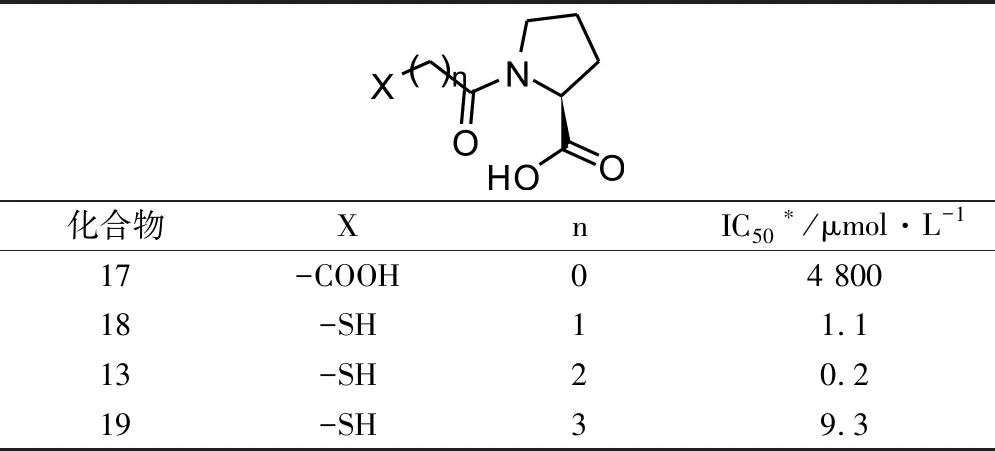
表3 不同脂肪链长度的活性数据
注:*使用的是兔血管紧张素转化酶
表3活性数据结果显示,当碳原子数为1和2时,活性较优,对体外酶抑制活性分别为1.1和0.2 μmol·L-1。而当碳原子数小于1或者大于2后,化合物活性显著降低。因此,研究人员继续以化合物13作为先导化合物,进行结构优化。
1.3.4 脂肪链上取代基优化 由图2可知,化合物的9位与ACE活性位点之间可能存在相互作用。另外,结合底物Ⅰ的结构,发现该位点之前有甲基取代。于是,研究人员在脂肪碳链的8位和9位分别引入甲基,继续研究化合物的构效关系(见表4)。
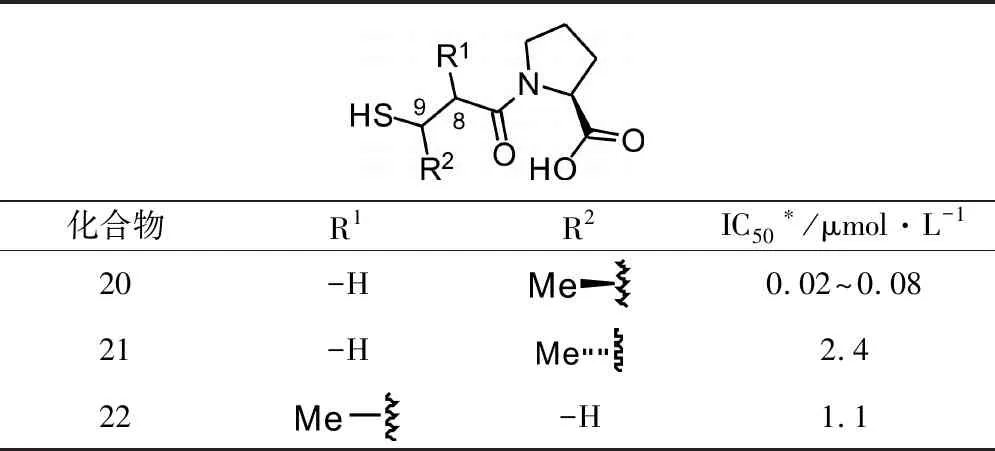
表4 脂肪链上引入不同取代基的活性数据
注:*使用的是兔血管紧张素转化酶
表4活性测试结果表明,当9位有甲基取代,化合物22活性降低。当8位引入甲基,S构型的化合物21较化合物13活性降低;但是,R构型的化合物20较化合物13活性提高了10倍左右。说明8位引入R构型的甲基更易与ACE的活性位点相互作用。
1.4 卡托普利上市及后续进展 最后,研究人员开展了化合物20(卡托普利)对肾脏高血压大鼠和正常血压大鼠的体内药效学实验(见图4),发现卡托普利对肾高血压大鼠具有非常好的降压效果,而对正常血压大鼠并没有明显影响[5]。再结合之前不同衍生物对ACE酶水平的抑制活性研究,研究人员正式将卡托普利作为候选药物,并于1977年全面开始临床研究,1981顺利获得FDA批准上市,成为首个上市的普利类ACEI[16]。
随着默沙东研发团队对卡托普利上市后的持续关注,发现巯基的引入带来了强疗效的同时也带来了许多副作用,例如皮疹、瘙痒以及味觉障碍等。于是,研究人员将卡托普利上的巯基直接用羧基替换,再对结构进行优化,最后发现用α-羧基苯丙胺取代时活性最好,其改善了与活性中心的亲和力,即依拉普利拉(Enalaprilat)。但是,依拉普利拉的口服生物利用度太差,半衰期短(T1/2=0.5 h,F%<10%)。研究人员通过前药设计的概念,将依拉普利拉的羧基做成单乙酯,提高了脂溶性,也就是现在的依拉普利(Enalapril)(二甲酸类),并在1985年被FDA批准上市。依拉普利具有较优的透膜性,易吸收(T1/2=11 h,F%=68%),在体内水解成羧酸再游离出来,从而发挥降血压的作用,成为继卡托普利后又一治疗高血压的重磅药物。随着药物化学家们对普利类药物的继续研究,又相继设计了许多不含巯基的二甲酸类药物,包括如:雷米普利(Ramipril)、贝那普利(Benazepril)等新产品。这些普利类药物都表现出优良的降血压效果,为高血压病人战胜疾病带来了福音。
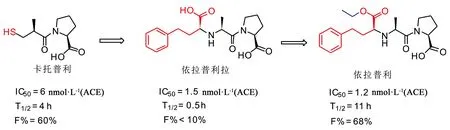
图4 从卡托普利到其他普利类药物的开发过程
2 总结
纵观卡托普利的研发历程可谓是一路波折,最初从蛇毒中发现导致低血压的缓激肽,再到从蛇毒中分离出缓激肽的稳定剂九肽活性化合物teprotide。临床试验发现teprotide的确是有效的ACEI,由于它是一种肽类分子,口服生物利用度低,限制了其在临床上的应用。然后,研究人员以化合物teprotide为基础,探讨ACE与其抑制剂可能的结合模式,设计出先导化合物1;进一步对先导化合物1的5个结合位点官能团的结构优化,成功得到了第一个口服类小分子ACE抑制剂卡托普利(Captopril)。卡托普利的研发过程汇聚了药物学家们的智慧与汗水,其成功经验对肽类苗头化合物的研发具有重要参考价值。
