HPLC波长切换法同时测定北芪五加片中7种成分的含量
2020-03-10钱玉梅孙艺爽
钱玉梅,孙艺爽
(1.河南省肿瘤医院 药学部,河南 郑州 450008;2.武汉大学 基础医学院,湖北 武汉 430071)
北芪五加片收载于中国药典2015年版一部[1],是由黄芪和刺五加浸膏加工而成的中成药复方制剂,具有益气健脾、宁心安神的功效,临床多用于心脾两虚、心神不宁所致的失眠多梦、体虚乏力、食欲不振等病症的治疗。现行质量标准中仅以黄芪甲苷作为质量控制指标,未对处方中其他成分进行定量检测,单一成分难以全面反映北芪五加片的整体质量,文献中也仅检索到对该制剂中紫丁香苷的含量检测[2]。中成药复方制剂其临床治疗效果往往都是多个有效成分共同作用的结果,多指标控制模式已逐步应用于中成药复方制剂的质量控制。该制剂方中黄芪为豆科植物蒙古黄芪或膜荚黄芪的干燥根,性甘,味微温,具有补气升阳、固表止汗、生津养血等功效,主要含黄酮类、多糖类、皂苷类等成分,其中毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素等为其黄酮类主要成分,黄芪甲苷为其皂苷类主要成分,中国药典2015年版一部黄芪药材项下以黄芪甲苷和毛蕊异黄酮葡糖苷为其控制指标;方中刺五加浸膏为五加科植物刺五加的干燥根和根茎或茎经加工制成的浸膏,中国药典2015年版一部刺五加浸膏项下以紫丁香苷、刺五加苷E和异嗪皮啶为其控制指标。本试验首次采用高效液相色谱(HPLC)波长切换技术,同时对北芪五加片中毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的含量进行测定,所建立的方法准确、简便、灵敏,为北芪五加片质量标准的提高和完善提供有力的实验基础。
1 仪器与材料
1.1 仪器
Agilent 1200型高效液相色谱仪(美国Agilent公司);AG285型电子分析天平(瑞士Mettler-Toledo公司);KQ-400KDE型超声波清洗器(昆山超声仪器有限公司)。
1.2 材料
毛蕊异黄酮葡糖苷对照品(批号:111920-201606,含量97.6 %),芒柄花素对照品(批号:111703-201504,含量100.0 %),紫丁香苷对照品(批号:111574-201605,含量95.2 %),刺五加苷E对照品(批号:111713-201804,含量97.9 %),异嗪皮啶对照品(批号:110837-201608,含量100.0 %,中国食品药品检定研究院);芒柄花苷(批号:486-62-4,含量95.0 %),毛蕊异黄酮(批号:20575-57-9,含量98.0 %,上海纯优);北芪五加片(规格:每片重0.5 g,批号:180805,181025,190127,山西晋新双鹤药业);乙腈为色谱纯,其他试剂为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱为Agilent TC-C18柱(4.6 mm×250 mm,5 µm),柱温为30 ℃;流动相为乙腈-0.1 %磷酸溶液,梯度洗脱(0~11.0 min,29.0 %A;11.0~34.0 min,29.0 %A→37.0 %A;34.0~49.0 min,37.0 %A→42.0 %A;49.0~55.0 min,42.0 %A→ 29.0 %A),流速为1.0 ml/min;检测波长分别为254 nm(0~34.0 min,检测毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素)[3-4]和220 nm(34.0~55.0 min,检测紫丁香苷、刺五加苷E、异嗪皮啶)[1,5];进样量为10 µl。
2.2 溶液的制备
2.2.1 对照品溶液 分别精密称取毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶对照品适量,用50 %甲醇制成每1 ml含毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶分别为0.676,0.932,0.894,2.736,0.598,0.352,0.138 mg的单成分对照品储备溶液;再分别精密吸取上述单成分对照品储备溶液各2.5 ml,用50 %甲醇制成混合对照品溶液(各对照品浓度分别为:毛蕊异黄酮葡糖苷33.8 mg/L、芒柄花苷46.6 mg/L、毛蕊异黄酮44.7 mg/L、芒柄花素136.8 mg/L、紫丁香苷29.9 mg/L、刺五加苷E17.6 mg/L、异嗪皮啶6.9 mg/L)。
2.2.2 供试品溶液 取北芪五加片适量,除去薄膜衣,研细,取1.0 g,精密称定,加50 %甲醇25 ml,密塞,称重,超声提取30 min,放冷,50 %甲醇补重,摇匀,过滤,制成北芪五加片供试品溶液,备用。
2.2.3 阴性样品溶液 按中国药典2015年版一部北芪五加片项下的处方比例及工艺,分别制备缺黄芪、缺刺五加浸膏的阴性样品,按2.2.2项方法操作,制成两种阴性样品溶液。
2.3 系统适应性试验
分别精密吸取2.2项下制备的4种溶液各适量,按2.1项色谱条件进样检测,记录色谱图。结果显示所记录色谱图基线平稳,北芪五加片中所测定成分毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶与其他峰均能达到有效分离,分离度均大于1.5;理论塔板数按所测定成分色谱峰计均大于4000;阴性样品在所测各成分的保留时间处无色谱峰干扰,色谱图见图1。
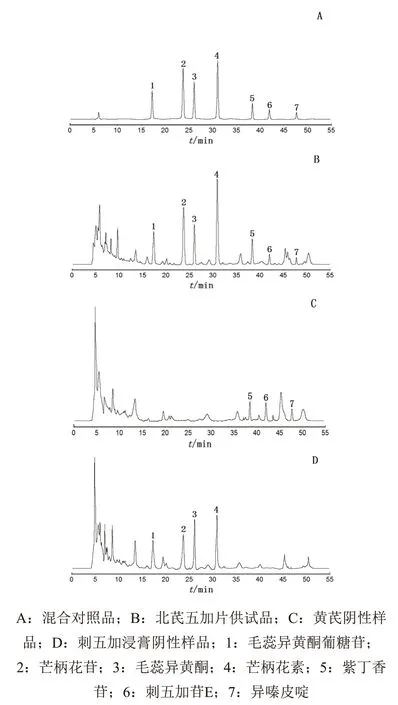
图1 北芪五加片HPLC色谱图
2.4 线性关系考察
分别精密吸取2.2.1项下单成分对照品储备溶液各0.1,0.2,0.5,1.0,1.5,2.0 ml,用50 %甲醇稀释成系列混合对照品溶液,依法进样检测,以毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶峰面积y为纵坐标,质量浓度x为横坐标,绘制标准曲线,得回归方程,以10倍信噪比计算各待测成分的定量限(LOQ),结果见表1。

表1 线性关系试验结果
2.5 精密度试验
取2.2.1项下混合对照品溶液,连续进样6次,记录毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的峰面积。结果所测定成分峰面积的RSD分别为0.72 %,0.66 %,1.61 %,0.54 %,1.12 %,0.89 %和1.31 %。
2.6 重复性试验
取同一批次北芪五加片样品,按2.2.2项下方法平行制备供试品溶液6份,依法测定测得毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的峰面积,计算所测定成分含量。结果毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的平均含量分别为0.885,1.221,1.031,3.679,0.718,0.391,0.148 mg/g,RSD分别为1.17 %,1.08 %,0.25 %,1.24 %,1.73 %,0.86 %和1.92 %。
2.7 溶液稳定性试验
取同一份北芪五加片供试品溶液,于0,2,4,8,16,24 h依法进样测定,记录毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的峰面积。结果北芪五加片供试品溶液24 h内稳定(室温),毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶峰面积的RSD分别为0.69 %,0.73 %,1.59 %,0.56 %,1.08 %,1.72 %和1.29 %。
2.8 加样回收率试验
精密称取毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶峰对照品适量,用50 %甲醇制成每1 ml分别含上述对照品0.446,0.608,0.522,1.836,0.352,0.198,0.074 mg的加标用混合对照品溶液。取同一批次已知含量的北芪五加片9份,除去薄膜衣,研细,每份0.5 g,精密称定,置具塞锥形瓶中,分别精密加入加标用混合对照品溶液0.5 ml 3份、1.0 ml 3份、1.5 ml 3份(对照品加入量约相当于样品含有量的50 %,100 %,150 %),按2.2.2项方法制成加样回收样品溶液,依法进样检测毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的峰面积,计算回收率及RSD值,结果见表2。
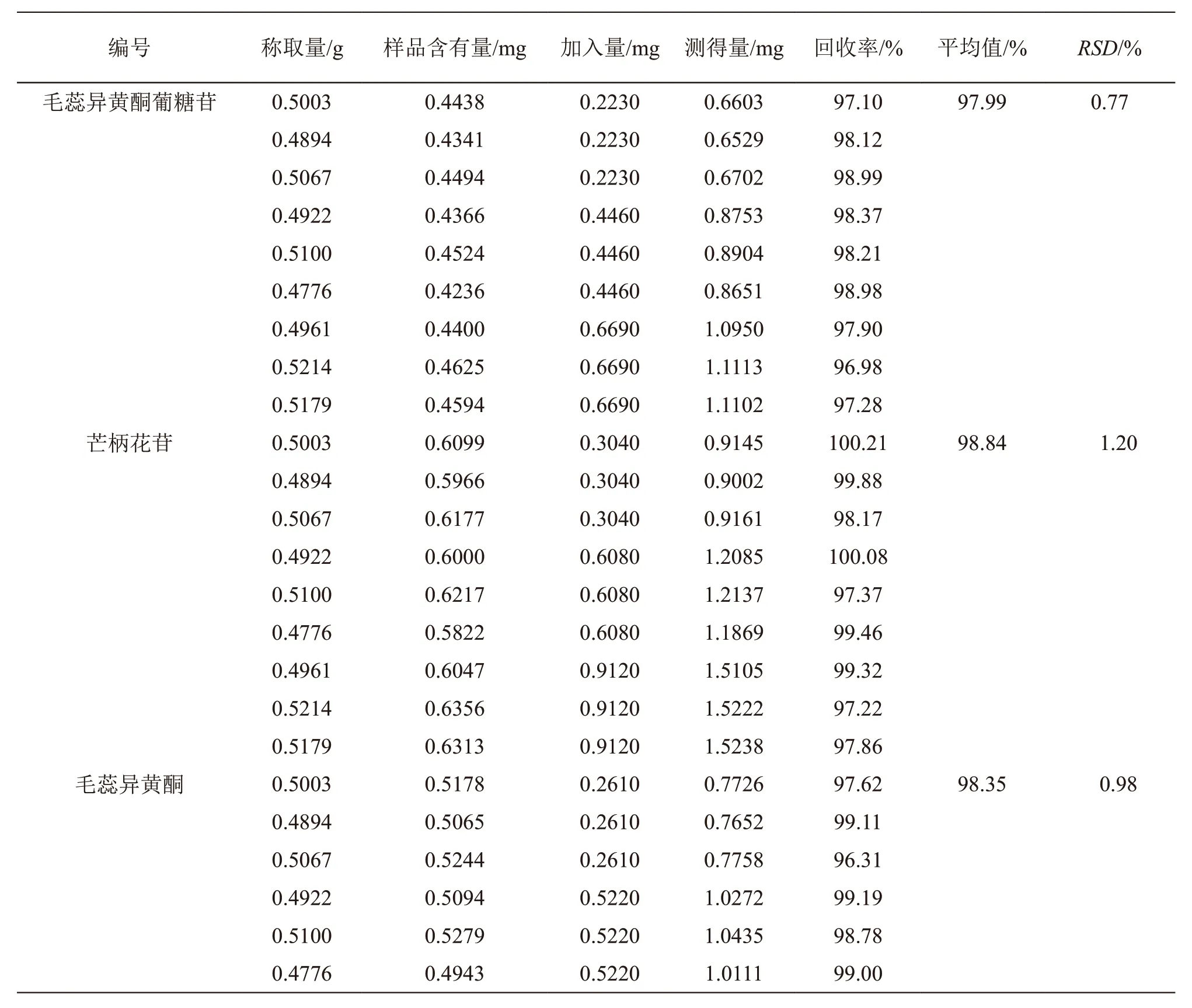
表2 毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的回收率试验结果
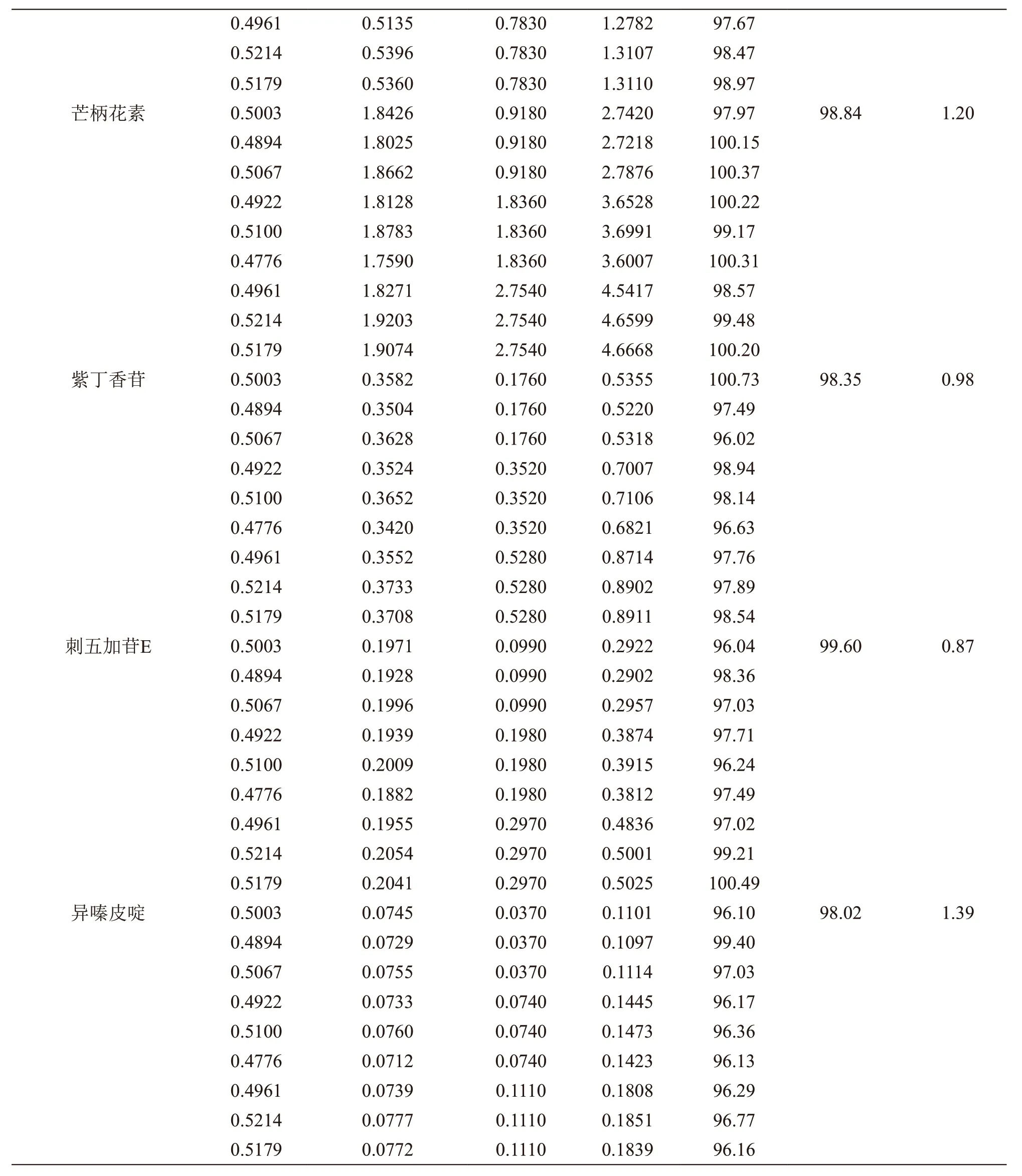
续表接上页
2.9 样品测定
取3个批次的北芪五加片,分别按2.2.2项下方法每个批次平行制备3份供试品溶液,按2.1项下色谱条件检测毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的峰面积,计算测定成分的含量,结果见表3。
3 讨论
3.1 北芪五加片制备方法的优化
试验中首先考察提取溶剂对毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶提取率及色谱峰峰形的影响。在超声提取60 min的同一条件下,选取甲醇[1,3-4,6]、50 %甲醇[1-2,5,7]、乙醇、50 %乙醇为提取溶剂,结果以50 %甲醇为提取溶剂时,毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶综合提取率最佳,同时待测成分色谱峰峰形较好;采用50 %甲醇为提取溶剂,对比了超声提取[1-7]、加热回流提取[8]、微波辅助提取的提取效率,结果3种提取方式下待测定成分毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶综合提取率差异不大,考虑到供试品溶液制备的便捷性和试验操作的可普及性,选取超声提取作为北芪五加片供试品溶液的制备方式;同时对超声时间进行了进一步摸索,最终确定采用50 %甲醇超声提取30 min用于制备北芪五加片供试品溶液。
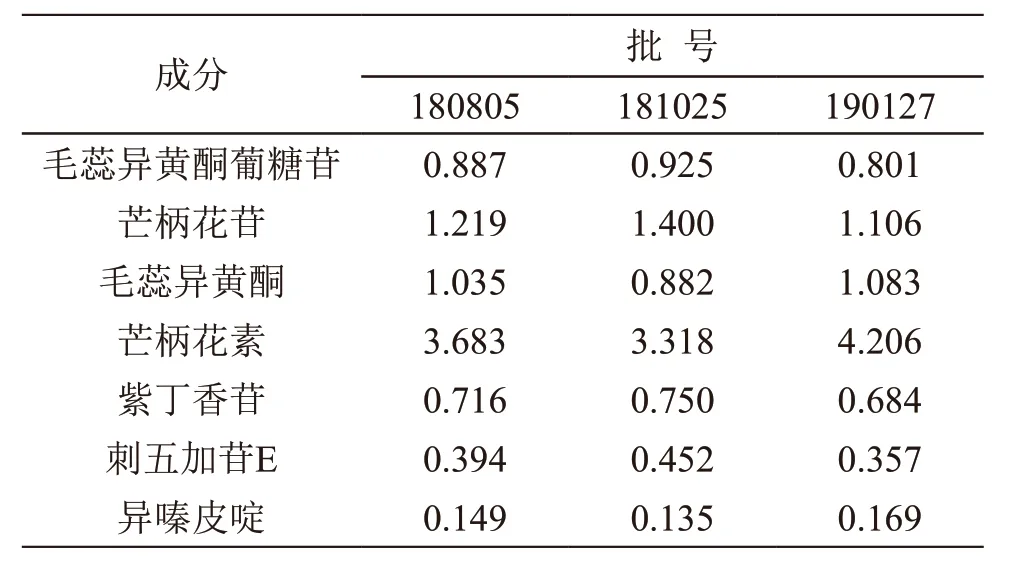
表3 含量测定结果(n=3,mg/g)
3.2 流动相的优化
在确定流动相时,首先考察了乙腈-水[4,8]、甲醇-水[2]流动相系统,结果发现以甲醇-水为流动相时待测定成分芒柄花苷、芒柄花素达不到基线分离,以乙腈-水为流动相时待测成分毛蕊异黄酮、紫丁香苷、异嗪皮啶峰存在拖尾;对此作者又考察乙腈-0.1 %甲酸溶液[1,3]、乙腈-0.1 %磷酸溶液[1,5-6]流动相系统,最终确定采用乙腈-0.1 %磷酸溶液为流动相,按2.1项下流动相比例对待测成分毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶进行梯度洗脱,在该流动相条件下,所记录色谱图基线平稳,待测成分毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶峰形对称,分离度符合规定要求。
3.3 检测波长的选择
在检测波长的选择过程中,分别取待测成分对照品溶液在200~400 nm范围内进行扫描,结果毛蕊异黄酮葡糖苷在219,254 nm和290 nm处有较大吸收,毛蕊异黄酮在218,253 nm和291 nm处有较大吸收,芒柄花苷在255 nm和301 nm处有较大吸收、芒柄花素在254 nm和300 nm处有较大吸收,紫丁香苷在220 nm和264 nm处有较大吸收,刺五加苷E在221 nm和272 nm处有较大吸收,异秦皮啶在223 nm和341 nm处有较大吸收,同时参考相关文献[1,3-5],最终确定采用波长切换法,毛蕊异黄酮葡糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素的检测波长为254 nm,紫丁香苷、刺五加苷E、异嗪皮啶的检测波长为220 nm,在此波长条件下,待测成分色谱峰分离效果较好,峰形较佳,杂质峰干扰少,灵敏度较高。
本研究首次建立一种HPLC波长切换法同时测定北芪五加片中毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、紫丁香苷、刺五加苷E、异嗪皮啶的含量,方法简便,准确,重现性好,可实现对北芪五加片多种有效成分的质量控制,为其质量标准的提高提供了可靠的数据依据。
