原始细胞低于20%伴RUNX1-RUNX1T1阳性AML 1例报道
2020-03-03李菁原叶向军卢兴国吴婧妍
李菁原, 叶向军, 卢兴国, 陈 燕, 吴婧妍, 彭 媛
(1.杭州迪安医学检验中心骨髓室,浙江 杭州 310000;2.兰溪市人民医院,浙江 兰溪 321100 )
急性髓细胞性白血病(acute myeloid leukemia,AML)的骨髓或外周血原始细胞一般需≥20%,但是伴t(8;21)(q22;q22.1);RUNX1-RUNX1T1(AML1-ETO)、inv(16)(p13.1q22)或 t(16;16)(p13.1;q22);CBFB-MYH11和PML-RARA的AML,原始细胞比例可以低于20%,但临床上少见。本研究报道1例原始细胞<20%伴RUNX1-RUNX1T1(AML1-ETO)阳性的AML病例。
1 病例资料
患者,男,75 岁,因头昏乏力半月余和血常规检查异常1周,疑诊白血病或骨髓增生异常综合征(myelodysplastic syndrome,MDS)入院。
血常规检查:白细胞计数8.06×109/L,原始细胞 7%(偶见Auer小体),早幼粒细胞8%,中幼粒细胞41%,晚幼粒细胞11%,中性杆状核粒细胞2%,中性分叶核粒细胞1%,淋巴细胞27%,单核细胞2%;血红蛋白47 g/L,红细胞计数1.54 ×1012/L,血小板计数6×109/L。幼粒细胞胞质大多缺乏嗜苯胺蓝颗粒,胞质呈浅杏红色一片。见图1。
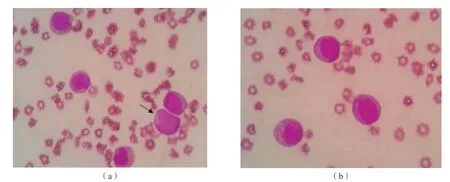
图1 本例患者外周血涂片
骨髓检查:有核细胞增生活跃,粒系异常增生。细胞分类示原始细胞10%,早幼粒细胞7%,中幼粒细胞40%,晚幼粒细胞12%,杆状和分叶核粒细胞6%,有核红细胞6%。计数100个粒细胞,其中67%为嗜苯胺蓝颗粒缺少,伴胞质杏红色明显,8%为少分叶核粒细胞和染色质凝聚异常。巨核细胞全片37 个,小圆核巨核细胞占 8%。见图2。
流式细胞术检测:以CD45/SSC设门,检测白血病免疫表型20种单抗,见图3。结果显示,原始细胞占有核细胞计数的4.6%,CD34、CD117、HLA-DR、CD13、CD56、CD38阳性,CD33、CD5、CD7、CD3、CD19、CD20、CD22等阴性,提示髓系原始细胞免疫表型;粒细胞比例升高(87.5%),CD13、CD16、CD11b表达减弱,提示幼稚粒细胞增多,且部分细胞异常表达CD56。
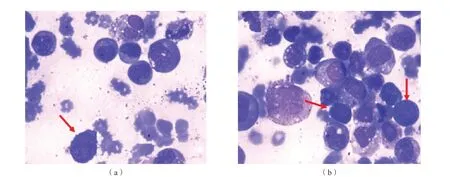
图2 骨髓象

图3 骨髓细胞流式免疫表型检查
白血病43种融合基因检测:RUNX1-RUNX1T1(AML1-ETO)阳性,其他融合基因均为阴性。常规染色体分析未检查。
2 讨论
t(8;2l)(q22;q22)是AML中最常见的染色体易位之一,预后较好,常见于法美英(French-American-British,FAB)分型中的M2。该易位导致位于8号染色体上的RUNX1T1(ETO)基因与21号染色体上的RUNX1(AML1)基因融合,形成了RUNX1-RUNX1T1融合基因。其产物融合蛋白通过与CBFβ形成异二聚体,与正常RUNX1产物负性竞争与DNA的结合,导致正常靶基因转录受抑制,引起髓系分化阻滞,使人体造血功能受损。融合蛋白还可导致TP53反应基因的激活,这可能部分解释了t(8;21)AML的相对化疗敏感性。融合基因虽然不足以引起白血病,但可形成自我更新能力更强的前白血病细胞群,随着时间变化演化出别的突变并发展为白血病,故有70%以上的患者可检出其他的遗传学异常[1-2]。融合基因RUNX1-RUNX1T1曾被称为AML1-MTG8和CBFα-ETO。
按世界卫生组织造血和淋巴组织肿瘤分类标准(2017 版),本型白血病被界定为有t(8;21)(q22;q22);RUNX1-RUNX1T1,而形态学常显示粒系细胞成熟特征和临床预后良好的AML,临床上多见于年轻人,初诊时可见髓系肉瘤(粒细胞肉瘤)和无法解释的骨髓原始细胞百分比降低。本例为老年患者,外周血涂片和骨髓涂片均显示原始细胞百分比较低,而以中幼粒细胞为主的幼粒细胞显著增加,并具有胞质颗粒缺少和着色浅红一片的特点。原始细胞大小不一,胞质特点为胞质量较丰富、嗜碱性并可见嗜苯胺蓝颗粒,与世界卫生组织分类规则中的描述基本一致。一些原始细胞还可见大肉色颗粒,被称为假性Chediak-Higashi颗粒,这被认为是颗粒的异常融合;Auer小体常见,为单一长形和锥体样,并可见于成熟中性粒细胞中。本例患者骨髓中早幼粒细胞和中幼粒细胞明显增多,且多为嗜苯胺蓝颗粒缺少及胞核异常[异常核分叶(如假性Pelger-Huet核)和/或胞质染色异常(如粉红色均匀性胞质)]的病态细胞(图 2)。流式免疫表型特征为原始细胞CD34、CD117、HLA-DR、CD13和CD56阳性,幼粒细胞表达模式异常,同时部分细胞表达CD56。幼粒细胞表达CD56可能是本型白血病的一个特点。有研究表明,在AML/ETO融合基因阳性的AML中,AML-M2占90%以上, 其他依次分别为AML-M4、AML-M1、AML-M5等,AML-M2中的大多数病例多为AML-M2b, 形态学上多表现为异常的中幼粒细胞比例增高[3]。有研究结果显示,CD56阳性者预后不良[4]。
本例患者若不做遗传学检查,则符合世界卫生组织的MDS-EB形态学标准,后者与AML的诊治明显不同。我们认为在形态学检查中,当原始细胞比例不如其他类型高,细胞成熟特征明显,伴有异常(早)中幼粒细胞明显增高(我国的M2b)并有形态学特征(胞质颗粒缺少和浅红色一片)者,应高度疑似本型AML;即使原始细胞比例低于20%也应使用遗传学检测证实是否为本型AML。此遗传学异常在常规细胞遗传学中通常明显,但少数病例可有亚显微易位,建议采用荧光原位杂交或分子方法检测RUNX1-RUNX1T,尤其是RT-PCR方法可检出有些FISH不能检出的隐蔽易位,而且可用作微小残留病检测,灵敏度可达10-3~10-4[5]。本病例未做常规细胞遗传学检查,可能有若干信息的缺失,但从遗传学原理上分析,该亚型白血病的特征其实主要是RUNX1-RUNX1T融合基因。该融合基因阳性,细胞遗传学检查t(8;21)易位阴性者可以为正常核型、复杂核型(涉及第8、第21和第3条染色体)或第8条染色体的缺失或其他异常,其具有相同的疾病特征;另从临床实践来看,常规细胞遗传学检查因阳性率低、报告时间长,正逐步被检测快速的分子技术替代,除了本例的AML伴RUNX1-RUNX1T外,临床实践中的APL伴PML-RARA亦是如此[6]。因此,分子遗传学技术可以帮助未进行细胞遗传学检查或细胞遗传学检查阴性者作出诊断[7]。
