视神经脊髓炎谱系疾病合并重症肌无力的临床特点及相关机制
2020-02-19刘静怡谭颖徐雁管宇宙朱以诚彭斌崔丽英
刘静怡 谭颖 徐雁 管宇宙 朱以诚 彭斌 崔丽英
视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)是一种免疫介导的中枢神经系统炎症性脱髓鞘性疾病,多认为由水通道蛋白-4抗体(aquaporin-4 antibodies,AQP4-Ab)与位于星形胶质细胞足突上的AQP4选择性结合,诱发抗体依赖及补体介导的细胞毒性反应等过程而致病。重症肌无力(myasthenia gravis,MG)是最常见的神经肌肉疾病之一,其涉及的一系列抗体与神经肌肉接头处的乙酰胆碱受体(acetylcholine receptor,AChR)、肌肉特异性激酶(muscle-specific kinase,MuSK)、脂蛋白受体相关蛋白4(LRP4)或其他突触后蛋白进行结合,造成神经肌肉信息传递紊乱,导致其特异临床表现。
近年来,NMOSD与其他自身免疫性疾病间的关系越来越受到关注。虽然较为少见,但临床仍可观察到部分NMOSD与MG共存的情况。目前尚不清楚此两疾病间的关系,但对此种共病的了解,可能为其诊断、治疗及预后带来有利影响。本文总结作者医院收治的2例NMOSD合并MG患者的临床特点并进行文献复习,初步对相关机制进行分析。
1 对象和方法
1.1 对象
1.1.1病例报告:2例NMOSD合并MG患者来自北京协和医院炎性脱髓鞘疾病临床研究队列(MSNMO Base)。自2011年1月至2019年4月,该队列研究共入组654例NMOSD患者,均符合2015年NMOSD诊断标准。本研究通过北京协和医院伦理委员会批准,经患者知情同意后获得临床资料。
病例1患者男,35岁。2011年1月无明显诱因突发左下肢麻木、无力,排尿费力,半月后肢体麻木无力自行改善,排尿费力持续。2012年2月突发胸部以下麻木、束带感,双下肢无力,伴便秘,就诊于作者医院,行头MRI未见病灶,颈胸椎MRI示T2~T5节段异常信号。2012年5月患者胸部以下麻木及双下肢无力明显加重,并出现复视。脑脊液检查显示,白细胞14×106/L(单核为主),蛋白0.58 g/L。血AQP4-Ab(+),ANA-IgG (+,1∶80),抗SSA(+++,129),抗Ro 52 (++,67)。既往史:2008年无明显诱因出现眼睑下垂、复视,疲劳可加重,休息可改善,晨轻暮重,于外院行新斯的明试验阳性,胸部CT未见明显胸腺异常,诊断MG,予以激素口服(具体用法不详),后长期口服泼尼松10 mg 1次/d,症状明显改善,否认面肌、延髓肌、呼吸肌无力及疲劳不耐受表现。入院查体:双眼右视可见粗大水平眼震,可引出复视,双下肢肌力5-级,左侧腱反射较右侧活跃,双侧Babinski征阳性,双侧T4水平以下针刺觉及音叉振动觉减退,走直线不稳,Romberg征阳性。行头MRI检查见右侧中脑、脑桥、桥臂多发斑点状异常信号(图1)。经口腔科及眼科会诊,并行干燥综合征(SS)相关眼部及涎腺检查,考虑未达到SS诊断标准。结合其症状、体征及实验室检查,诊断NMOSD合并MG。给予激素冲击后序贯减量,并加用硫唑嘌呤治疗,患者症状改善,出院查体示感觉平面下降(双侧T6~T7以下针刺觉减退,双髂以下音叉振动觉减退)。患者定期于作者医院门诊复诊,病情稳定,无明显MG及NMOSD复发表现。
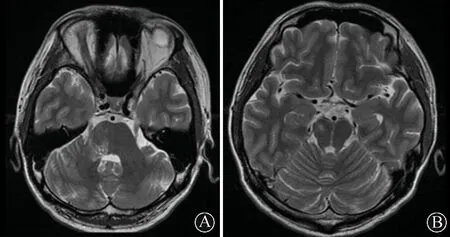
图1 病例1头MRI检查(2012年5月),可见右侧脑桥、桥臂(A)及右侧中脑(B)斑点状长T2异常信号
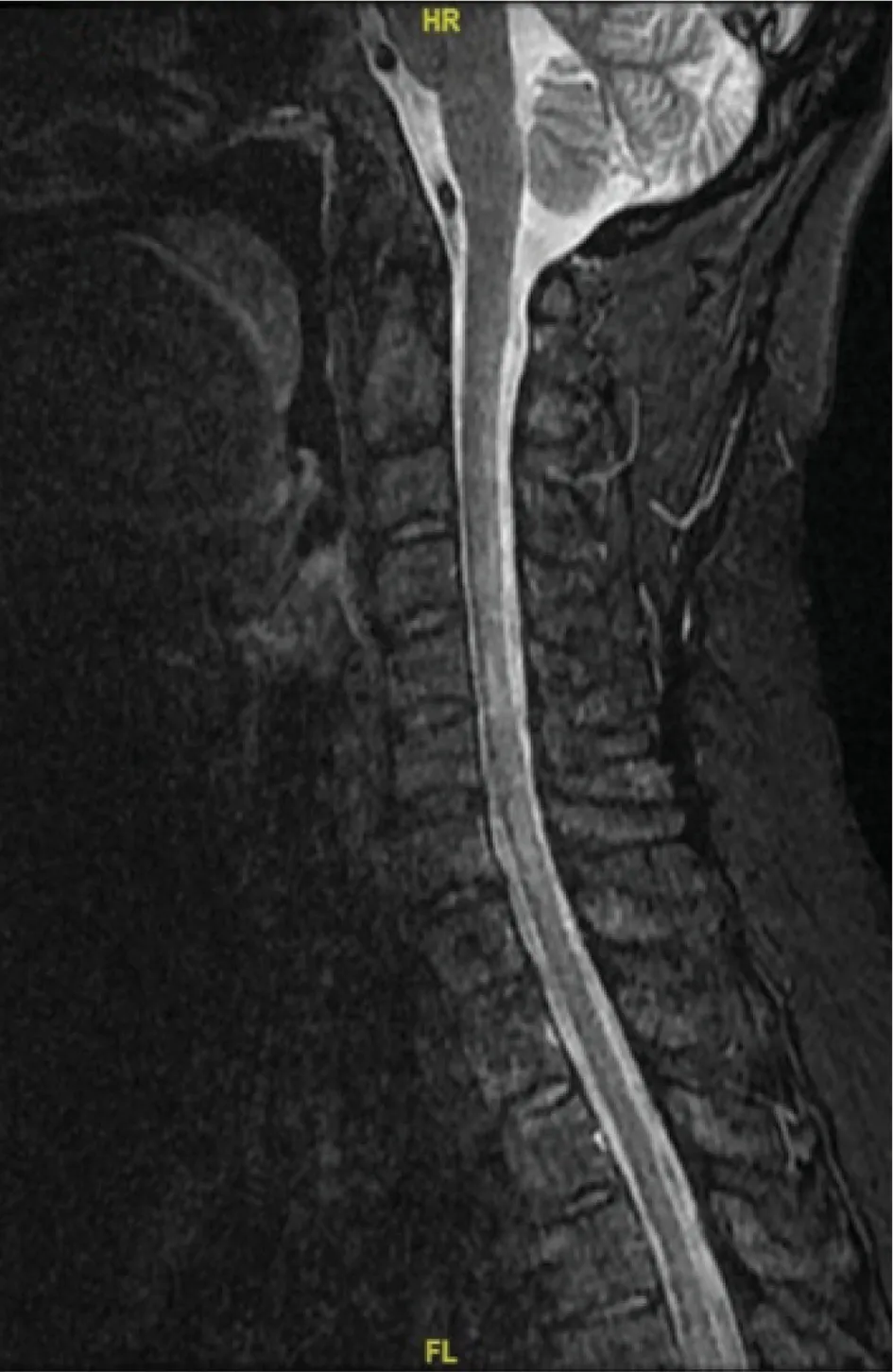
图2 病例2颈椎MRI检查(2014年4月)可见C1~C7长节段异常高信号
病例2患者女,48岁。2014年1月无明显诱因出现左侧肢体及脐以下腹部麻木,伴后颈部疼痛,行走踩棉感,行脊髓MRI检查提示C5及T5椎体水平脊髓异常强化影,于当地医院诊断“急性脊髓炎”,应用静脉注射免疫球蛋白(IVIg)联合激素冲击治疗后症状好转。2014年4月患者出现四肢麻木,伴胸部束带感,激素冲击治疗症状改善,后激素减量过程中出现躯干及四肢麻木无力,四肢间断痉挛伴疼痛,复查颈椎MRI见C1~C7髓内异常信号(图2),诊断NMOSD,经激素治疗后症状好转,仅遗留行走踩棉感,复查颈椎MRI见病灶缩小。2015年4月出现双下肢及双足麻木,迅速发展至躯干、左上肢、右面部及耳后,胸椎MRI检查示T4~T5水平髓内异常强化灶,经激素冲击序贯口服治疗后症状逐渐好转,仅遗留双肘以下及双足底麻木感。2015年10月出现视物不清,逐渐加重,间断言语不利,就诊于作者医院,查血AQP4-Ab(+)。视觉诱发电位(VEP)、脑干听觉诱发电位(BAEP)、视神经MRI检查结果正常,眼科考虑其视物不清为激素相关性青光眼可能。血常规检查提示血小板减低〔(12~28)×109/L〕,结合骨穿及血小板相关抗体等相关化验,血液科考虑Evans综合征可能性大,加用环磷酰胺0.4 g静脉滴注,1次/周。经治疗患者肢体麻木缓解,后规律口服泼尼松+静脉应用环磷酰胺,2016年6月停用环磷酰胺,改为口服吗替麦考酚酯0.75 g,2次/d。2017年4月患者出现间断右眼睑下垂及视物重影,疲劳后可有双手及双下肢乏力感,休息可好转,后频发夜间胸闷、疲惫感,就诊于外院,行新斯的明试验(+),诊断MG。予溴吡斯的明口服治疗,自觉症状明显改善。为系统诊治遂就诊作者医院,完善低频重复电刺激(RNS)检查示波幅递减。血AChR-Ab 3.87 nmol/L(+),AQP4-Ab 1∶32(+)。胸部CT检查未见胸腺瘤。既往史:甲亢病史20余年。结合患者临床表现以及实验室检查结果,诊断NMOSD合并MG。
1.1.2文献复习:以“neuromyelitis optica”[MeSH Terms]OR “Neuromyelitis optica spectrum disorder”[Text Word]OR“NMOSD”OR“NMO” AND “myasthenia gravis”[MeSH Terms]OR “MG”为检索指令,检索PubMed数据库,时间截至2019-09-01。通过阅读文献(包括综述及个案报道),及所涉及的相关参考文献,获取其中报告的所有临床确诊NMOSD与MG共病的病例资料,对于未描述诊断依据及临床资料叙述欠详细的病例予以排除。
1.2 方法分析本文收集的2例以及文献报道的NMOSD合并MG患者的临床特点。
1.3 数据处理采用SPSS 25.0软件建立数据库,并对数据进行汇总分析。
2 结果
共检索到24篇涉及NMOSD与MG共病患者的文献,其中共77例;结合本文报道的2例患者,共79例。NMOSD合并MG者以青中年为主,女性居多(64例,85.3%)。在有NMOSD及MG发病年龄或起病顺序明确记录的63例患者中,53例患者(84.1%)MG先于NMOSD发病。MG与NMOSD两病出现的间隔时间为0~25年,平均(12.05±5.04)年。79例患者中,共48例(60.8%)接受了胸腺切除术。NMOSD与MG共病者的MG类型多为全身型(55例,69.6%)。NMOSD与MG共病患者可合并其他免疫疾病〔系统性红斑狼疮(SLE)、SS等〕,及存在其他免疫相关抗体阳性(ANA、dsDNA、GAD-Ab等)。结果见表1。
3 讨论
本文2例合并MG患者来自北京协和医院炎性脱髓鞘疾病临床研究中的654例NMOSD患者,其发生率为3.06‰,远高于一般人群中MG患病率(0.15‰~0.25‰)。这与既往报道的结果趋于一致[2, 16, 22],提示MG和NMOSD的共存可能不仅仅是巧合。
既往研究显示,MG多先于NMOSD发生,且许多患者于出现NMOSD前曾行胸腺切除术。曾有研究对行胸腺切除术的MG患者行长期随访发现,其自身免疫性疾病的发生率为12.5%[25],提示胸腺切除术可能导致机体免疫失调,增加包括NMOSD在内的自身免疫性疾病的易感性。Chan等[26]对10例胸腺瘤患者(其中9例表现为MG)的胸腺瘤组织进行研究发现,无论临床是否表现为MG,胸腺瘤细胞膜均表达AQP4,而无胸腺瘤患者的胸腺组织则无AQP4表达。这表明在某些情况下,针对胸腺瘤细胞膜上表达的AQP4的免疫应答可能启动,从而继发NMOSD。这为NMOSD常发生于MG后的现象增加了一种可能解释,即这些患者中可能存在着常规影像学难以检测到的微胸腺瘤,这些微胸腺瘤表达AQP4,并在瘤体或胸腺组织中产生AQP4相关反应性T细胞,这种T细胞可从胸腺瘤或胸腺中输出,并驻留在胸腺周围淋巴结中,若在某些情况下被激活,则可在胸腺切除术后数个月或数年触发NMOSD。这种假说也可能为Leite等[6]发现的结果,即AQP4-Ab可在MG发病早期乃至NMOSD相关症状出现前数年被检测到提供解释。
但亦有一系列病例报道显示部分患者MG症状于NMOSD发病数年后出现,患者可无明显胸腺瘤或未经胸腺切除术,这与本文中报道的2例患者类似,表明MG、胸腺瘤或胸腺切除术并非是NMOSD发生的必要因素。因此,NMOSD与MG间的具体关联仍需进一步探讨。
NMOSD与MG具有许多相似之处:首先,两者的发生发展可能均与遗传因素相关,并受某些环境因素共同影响。部分人类白细胞抗原(human leukocyte antigen,HLA)基因型已被发现与MG及NMOSD有关[27],表明先天性免疫系统信号可能通过抗原呈递细胞(APCs)转化为特异性免疫反应,建立长期免疫记忆。在MG及NMOSD患者中均可观察到维生素D水平降低,维生素D水平与NMOSD疾病相关残疾、临床严重程度呈负相关,而MG患者经补充维生素D治疗后临床症状可获得改善[28-29]。其次,NMO及MG均依赖B细胞及T细胞介导的免疫过程,异常抗体的出现在两病中起关键作用:浆细胞分泌异常抗体,记忆B细胞产生促炎性细胞因子并加重自身免疫过程;研究表明Th17细胞计数百分比增加与AChR-Ab抗体滴度呈正相关[30],而NMOSD患者CSF及血清Th17相关的细胞因子及趋化因子水平常升高[31]。第三,从临床角度出发,两病均以女性多见,临床病程均有一定波动性,均需长疗程免疫调节治疗,均常与其他系统性免疫病(自身免疫性甲状腺疾病、SLE、类风湿关节炎、SS等)共存。另外,有研究表明,AQP4亦在神经肌肉接头处表达,可能作为两种疾病发生的共同靶点[32];而更多自身免疫抗体的不断发现,提示MG合并NMOSD患者可能存在其他暂未发现的具有双重病理机制的自身抗体。目前关于二者共病的治疗选择方面并无公认标准,在既往文献的病例报道中,亦缺乏对具体治疗方案及长期随访预后的相应描述。未来仍需更多的研究以揭示MG与NMOSD之间的关系,从而利于其治疗选择及预后改善。

表1 79例NMOSD与MG共病患者临床特点
注:NMOSD:视神经脊髓炎谱系疾病;MG:重症肌无力;AQP4-IgG:水通道蛋白4抗体;AChR-Ab :乙酰胆碱受体抗体;NK:未提及,后面数字表示例数;O=眼肌型MG,后面数字表示例数;G=全身型MG,后面数字表示例数;SLE:系统性红斑狼疮;SS:干燥综合征;APS:抗磷脂综合征;TPO:甲状腺过氧化物酶抗体;ANA:抗核抗体;GAD-Ab:谷氨酸脱羧酶抗体;a:于NMOSD发病前11~24年起病;b:于NMOSD发病前起病;c:中位数;d:14例发生于MG后;e:均发生于MG后;f:8例发生于MG之后
综上所述,MG与NMOSD可共存,两者发病机制可能存在一定联系。临床上当MG患者突然出现视神经、中枢神经受损表现,或当NMOSD患者出现疲劳性肌无力时,应考虑两病共存的可能性。在情况允许时,可对早发MG患者进行AQP4-Ab检测,当发现AQP4-Ab时需警惕其疾病进展的可能性。因此,对两病间联系的认识,将有助于临床诊断及疾病的科学管理。
