NanoSe-LMH-CS-SA复合载药微球的制备与缓释性能研究
2020-02-02吴永军刘红梅
吴永军,吉 民,刘 燕,刘红梅
1马鞍山师范高等专科学校 食品工程系,马鞍山 243041;2马鞍山丰原制药有限公司,马鞍山 243000
现有补硒制剂主要采用无机硒、有机硒作为硒源,存在毒性较大,吸收利用率低等一系列弊端,限制了补硒制剂的临床与日常应用[1]。相比而言,纳米硒(NanoSe)易于吸收利用,是己知硒制品中安全性最高的,无疑是补硒的最佳硒源[2]。
壳聚糖(chitosan,CS)是一种无毒的天然高分子多糖,其生物相容性好,无致突与致敏性,具有促进组织再生、调节免疫、降血压、降血脂及排毒等重要生理功能。壳聚糖作为一种聚阳离子电解质,结构上含有大量氨基,在酸性消化液中能膨胀成粘稠的胶体物质,有效阻滞包埋药物的溶出与扩散,在缓(控)释药物载体、靶向药物载体、骨架剂、成膜剂等领域具有广泛应用[3]。
海藻酸钠(sodium alginate,SA)是一种具有生物相容性、可降解的天然聚阴离子多糖,被广泛应用于缓释制剂的开发中。当壳聚糖与海藻酸钠混合时,壳聚糖分子链上的大量氨基与海藻酸钠的分子链上的大量羧基通过静电引力聚集成电解质膜,可有效包裹芯材、增强微球的稳定性、调节药物释放速度[4]。海藻酸是难溶性弱酸,沉淀pH值为3.6以下,溶解pH值为5.8,所以在进行微胶囊化过程中要调节混合液的pH值。
赖氨酸是谷物中的第一限制性氨基酸,以稻谷小麦为主食的膳食结构会导致我国居民赖氨酸摄入量不足,引发机体蛋白质代谢障碍,进而出现一系列生长发育问题[5]。L-盐酸赖氨酸(L-lysinemonohydrochloride,LMH)易溶于水,且属于GRAS物质,可用于人体日常补充赖氨酸。
纳米药物(制剂)是以微纳米级高分子材料为壁材,以一定包埋方式负载纳米级药物芯材,制成具备微球、微囊、胶束等特殊结构的新型药物(制剂);其具有缓控释放、靶向增浓、提高药物生物相容性及吸收利用率等突出优势[6,7]。本研究以带有相反电荷的壳聚糖、海藻酸钠作为壁材,吸附包裹NanoSe芯材,添加LMH作为复合营养物,CaCl2做交联剂,使用复凝聚法制备了NanoSe-LMH-CS-SA复合载药微球(简称复合载药微球);实验测试了壁材比、芯壁比、交联温度、交联剂用量对包埋率的影响,进而通过BBD设计优化微球制备工艺条件;借助体外释放实验测试了复合载药微球在两种不同模拟消化液中的硒释放率,以期为缓释型补硒制剂的研究与开发应用提供参考。
1 材料与方法
1.1 材料与仪器
壳聚糖(BR,脱乙酰度>85%,美国Sigma试剂公司);海藻酸钠(AR,成都西亚化工股份有限公司);LMH(BR)、CaCl2(AR)、CH3COOH(AR)、HCl(AR)、NaHCO3(AR)、NaOH(AR)、无水酒精(AR)(国药集团上海化学试剂有限公司);KH2PO4(AR,天津博迪化工有限公司);NanoSe溶胶(10.9 mg/mL,粒径20~50 nm,pH=3.0±0.5,烟台佳隆纳米产业有限公司);蒸馏水。
S-4800扫描电镜(日本Hitachi);LS-13-320激光粒度分析仪(美国贝克曼库尔特);Nicolet6700傅立叶红外光谱仪(美国尼高力);DTG-60H热重-差热分析仪(日本岛津);Vario EL III元素分析仪(德国元素分析系统公司);TU-1901紫外可见分光光度计(北京普析);BPZ-6210-2B真空干燥箱(上海一恒);DZTW-500型恒温电热套磁力搅拌器(上海力晨);TS-110X50恒温水浴摇床(上海天呈);FA1104型分析天平(上海舜宇恒平);TGL-16C型离心机(常州中捷);PHS-3C电子pH计(上海雷磁)
1.2 实验方法
1.2.1 复合载药微球制备
以1.0%乙酸溶液100 mL先后溶解1 g壳聚糖及1g CaCl2,配制成的壳聚糖与CaCl2浓度均为10 mg/mL的混合溶液,记为A液;同时,以1.0%海藻酸钠溶液(pH 5.8)100 mL溶解0.5 g LMH,配制5 mg/mL的LMH溶液,再以此LMH溶液稀释定量NanoSe溶胶,1 000 rpm均质10 min,记为B液。
用带8号针头的50 mL注射器吸取40 mL B液,自30 mm高度缓慢滴至10 mL A液中(每滴入20 mL B液调整一次滴加高度),使用HCl、NaHCO3溶液调节pH=5.0,300 rpm转速下40 ℃恒温交联60 min,离心分离沉淀,用蒸馏水清洗沉淀3遍,40 ℃真空干燥得到NanoSe-LMH-CS-SA复合载药微球。
1.2.2 复合载药微球结构表征
NanoSe-LMH-CS-SA复合载药微球的形貌使用SEM电镜进行检测,微球粒径分布使用激光粒度分析仪检测,微球的有机结构采用傅立叶红外光谱仪分析,微球的热效应采用热重-差热分析仪分析。检测条件如下。
1.2.2.1 形貌检测
SEM采用Hitachi S4800型扫描电镜,复合载药微球样品用酒精分散滴于铜片上并经喷金处理,二次电子检测器成像,加速电压5 kV,工作距离9 mm,对应放大倍率为22 000倍;工作距离9.1 mm,对应放大倍率为40 000倍。
载药微球粒度分布检测采用贝克曼库尔特LS-13-320激光粒度分析仪,检测条件为:固体激光器功率5 mW,检测波长780 nm,湿法检测、SOP模式,红光折射率1.82,吸收率0.1。
1.2.2.2 红外光谱检测
红外光谱分析采用美国尼高力Nicolet6700傅立叶红外光谱仪,检测条件为:光谱范围4 000~500 cm-1,光谱分辨率0.8 cm-1,噪音峰-峰值1×10-5Abs,KBr压片。
1.2.2.3 热稳定性检测
微球热稳定性分析采用日本岛津DTG-60H热重-差热分析仪,检测条件为:N2氛,气体流速30 mL/min,升温速率10 ℃/min。
1.2.2.4 元素组成分析
C、H、N元素分析使用Vario EL III元素分析仪,检测条件为:高纯O2压力0.20 MPa、流速10 mL /min,高纯He压力0.12 MPa、流速200 mL/min;燃烧炉温度1 150 ℃,还原炉温度850 ℃,样品以锡纸包裹进样。Se元素含量检测使用邻苯二胺紫外分光光度法[8],平行测定三次取均值。
1.2.3 体外释放单因素实验
自配硒标准溶液,采用邻苯二胺紫外分光光度法于335 nm波长下测硒标液样品吸光度A,以未加入复合载药微球模拟消化液为空白对照,绘制硒标准曲线[2]。
准确移取复合载药微球微球10 mg至筒状透析袋内,用透析夹固定两端浸于100 mL 2种不同pH值模拟消化液中,以HCl溶液(pH=1.0)作为模拟胃液,以磷酸盐缓冲溶液(pH 6.8)作为模拟小肠液。37 ℃恒温水浴摇床震荡,开始实验后间隔一段时间取袋外消化液1 mL,以邻苯二胺紫外分光光度法测定其吸光度、折算硒含量,并补充等量的模拟消化液,平行测得3次取均值,计算以下指标。
(1)

(2)

(3)
借助单因素实验考察壁材比、芯壁比、复凝pH值、交联剂用量对复合载药微球包埋率的影响。
1.2.3.1 壁材比对包埋率的影响
1.2.3.2 芯壁比对包埋率的影响
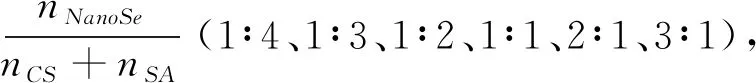
1.2.3.3 CaCl2用量对包埋率的影响
依据“1.2.1”制备流程,在条件实验确定的壁材比和芯壁比下,改变交联剂CaCl2用量(0.25、0.5、0.75、1.0、1.25、1.5、1.75、2.0g),其余制备条件不变,制备复合载药微球样品。随后采用“1.2.3”体外释放实验方法,提取消化实验开始后1 h的消化液,测量微球在两种消化液中的包埋率。
1.2.3.4 复凝pH值对包埋率的影响
依据“1.2.1”制备流程,在条件实验确定的壁材比、芯壁比和交联剂CaCl2用量下,考虑到壁材壳聚糖只能溶解于酸性溶液[9],所以将复凝pH设定在弱酸性环境,使用HCl、NaHCO3溶液改变复凝pH值(3.5~4.0、4.0~4.4、4.5~5.0、5.0~5.5、5.5~6.0),其余制备条件不变,制备复合载药微球样品。随后采用“1.2.3”体外释放实验方法,提取消化实验开始后1 h的消化液,测量微球在两种消化液中的包埋率。
1.2.4 响应面优化复合载药微球制备条件
在“1.2.3”单因素实验基础上,采用BBD响应面设计实验,确定的最佳条件优化复合载药微球制备工艺,制备优化型复合载药微球,对比优化前后微球的包埋率和载药量;借助体外释放实验,测试优化型复合载药微球在两种模拟消化液中0.5~12 h的释放性能。
2 实验与分析
2.1 微球结构分析
2.1.1 微球形貌表征与粒径分析
复合载药微球样品的SEM如图1所示,图中微球分散良好、表面凹凸不平,部分微球表面具有不规则的凹坑和孔洞,使其具有较大的比表面积,且上述凹坑或孔洞还可成为内部药物扩散的通道。复合载药微球样品的激光粒度分析检测结果如图2所示,载药微球样品的中粒径D50为10.7 μm,大部分微球样品(D10~D90)分布在6.2~12.9 μm范围内。
A液中壳聚糖(CS)胶束带正电荷,B液中溶有的海藻酸钠(SA)胶束带负电荷,同时赖氨酸(Lys)的等电点PI=9.74,溶于B液(pH<7)的L-盐酸赖氨酸(LMH)电离后以阳离子形式存在。试验中,当少量B液缓慢滴入大量A液,混合溶液的黏度较低,两种带有相反电荷的壁材在CaCl2作用下会迅速完成交联、混凝、聚沉,形成小粒度微球[10]。
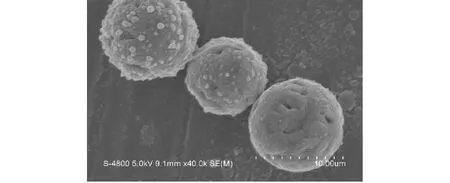
图1 复合载药微球样品的SEM形貌Fig.1 The SEM of drug-loading microspheres
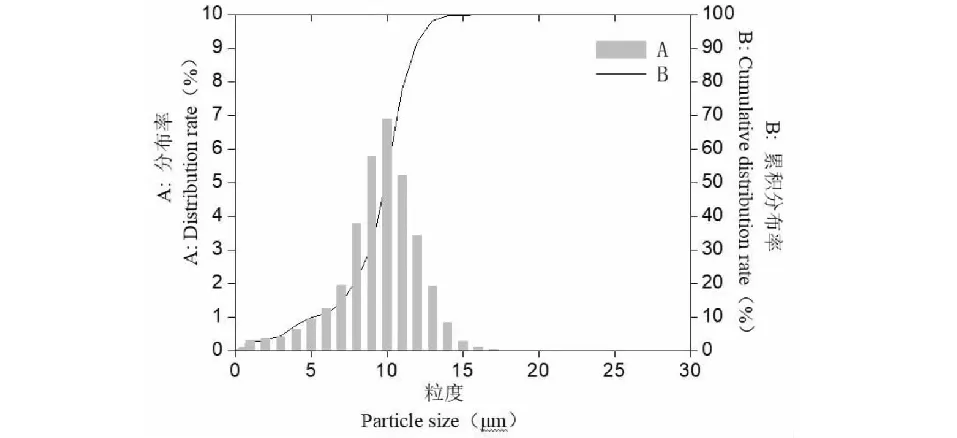
图2 复合载药微球样品的激光粒度分析Fig.2 Particle size analysis of drug-loading microspheres
2.1.2 红外光谱分析
如图3所示,CS的红外吸收图谱中,3 430 cm-1处的宽峰是-OH与-NH2的收缩振动多重耦合峰,1 640cm-1处是酰胺I谱带的羰基吸收和-NH2振动吸收的多重耦合峰。SA的红外吸收图谱中,3 430 cm-1处的宽峰是-OH的收缩振动峰,1 630 cm-1处是-COO-的收缩振动峰。LMH的红外吸收图谱中,2 940 cm-1处是C-H振动峰,1 622 cm-1处是-CO-的吸收峰,1 595cm-1和1 340 cm-1处是-COO-的不对称和对称收缩振动峰,1 550 cm-1处吸收峰以及低波数范围出现的上下波动可能是Cl-的影响,这在LMH标准IR谱中同样可见。
微球的红外吸收图谱中,两种壁材3 430 cm-1处的吸收峰在微球中没有改变,说明微球中SA和CS中原有的氢键结构没有改变;CS的1 640 cm-1处-NH2振动吸收峰和SA的1 630 cm-1处-COO-的收缩振动峰红移至1 622 cm-1处,与LMH的-CO-吸收峰合并,说明两种壁材复凝聚形成微球主要依靠CS上氨基与SA的羧基形成氢键。而LMH上1 595 cm-1和1 340 cm-1处-COO-的不对称和对称伸缩(收缩)振动峰在微球上没有出现,可能是其在微球形成过程中与壁材CS和SA中的-NH2和-OH形成了氢键所致,此时C-H受到壁材CS和SA中苯环或糖环的影响,C-H振动加强,由2 940 cm-1红移至2 926 cm-1。整体而言,微球与其主要组成的红外光谱在形态上大致相同。综合以上,能够证实两种壁材主要是以氢键和静电作用复凝聚形成复合载药微球。
2.1.3 热重-差热分析
复合载药微球样品及其组成材料的热稳定性分析如图4所示。图中微球的DTA-TGA图谱有别于微球组分CS、SA、LMH的DTA-TGA插图,微球与其组分的具有不同的热分解温度,说明CS、SA、LMH在复合载药体系中并非简单的混合,存在氢键和静电作用等结合力使得壁材复凝聚形成微球。
由微球的TGA曲线可见,微球的受热减重可分为“失水,脱羧,热分解”三个阶段。从室温加热至800 ℃,该微球样本的失重率为88.50%。其中:190 ℃以内,随着温度的升高,复合载药微球样品先后失去其中的物理和化学结合水,失重20.35%;190~480 ℃,微球中的CS-SA壳膜、LMH发生脱羧反应,其有机部分中羧基逐渐分解成CO2,致使微球样本快速失重45.01%。考虑到微球的结构,CS-SA壳膜可能先于LMH发生脱羧;480~525 ℃,微球有机部分CS、SA、LMH的碳骨架开始发生热分解,裂解出CO2、CO、H2O等,微球继续减重18.33%;525 ℃之后,微球失重不明显。由微球的DTA曲线可见,77 ℃处的吸热峰对应吸附水的脱除,位置与TGA曲线中失重第一台阶吻合;强放热峰出现的位置对应有机物热分解快速失重,其位置与TGA曲线中失重第三台阶吻合,分解温度为516 ℃。
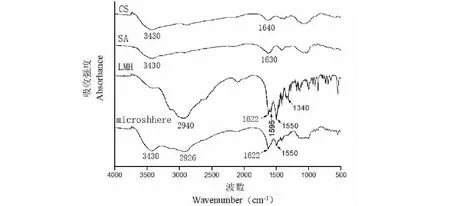
图3 复合载药微球样品及组分的的红外光谱Fig.3 The infrared spectrums of drug-loading microspheres and its components
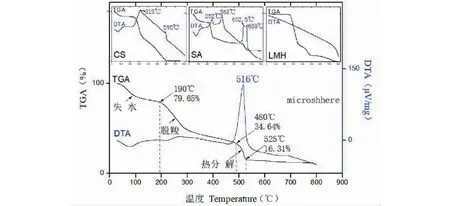
图4 复合载药微球样品及组分的TGA-DTA分析Fig.4 The TGA-DTA of drug-loading microspheres and its components
2.1.4 微球元素组成分析
微球及其组分的主要元素分析如表1所示,其中微球C、H、N、Se四种元素的含量分别为34.92%、6.53%、5.01%和2.47%。由于微球组分CS和SA、LMH中分别自带Na+和Cl-;同时,在复凝聚法制备微球过程中添加的CaCl2、混合磷酸盐会带入Ca、P等杂质;结合微球形成主要借助氢键及静电作用,故无法通过减量法计算O含量、进而推导微球可能的组成式。
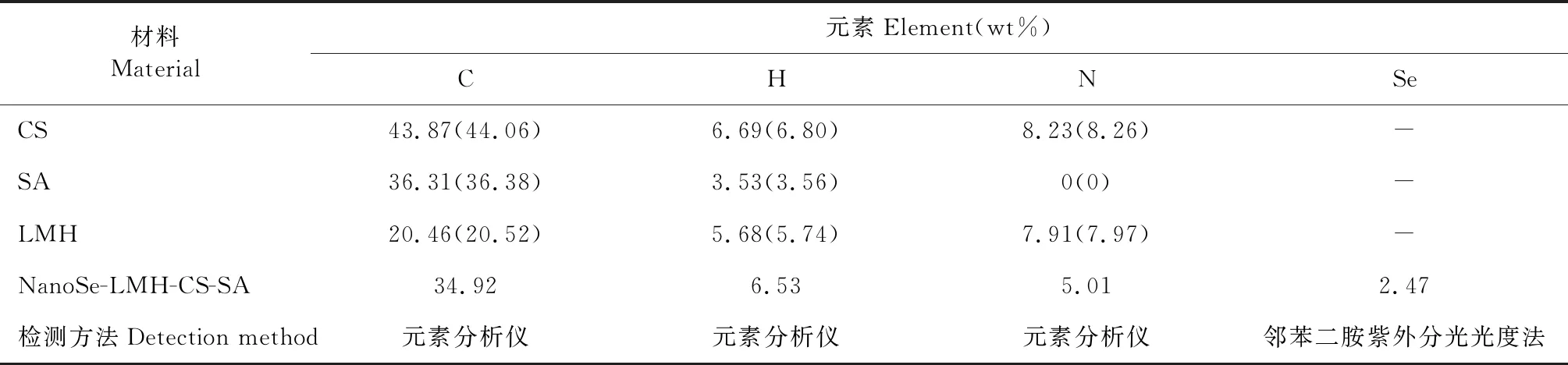
表1 复合载药微球与其组分的主要元素组成分析Table 1 Major elemental analysis of type II drug-loading microspheres and its components
2.2 体外释放单因素实验
2.2.1 壁材比对包埋率的影响
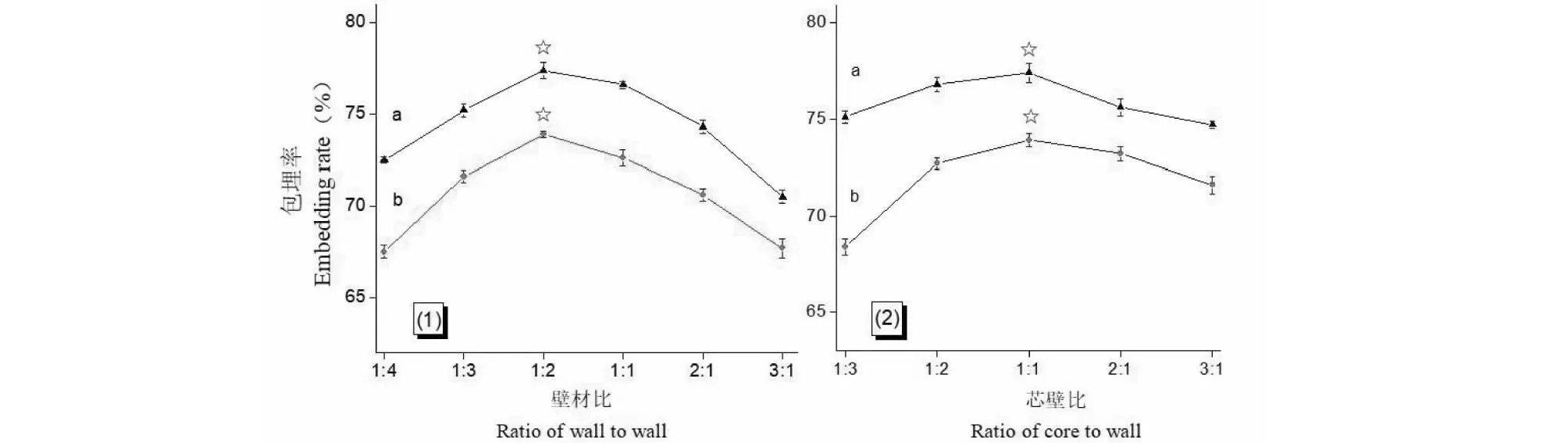
图5 壁材比、芯壁比对载药微球样品包埋率的影响Fig.5 The influence of ratio of wall to wall and ratio of core to wall to the embedding rate of microspheres注:a:模拟小肠液;b:模拟胃液,下同。Note:a:Simulated small intestinal juice;b:Simulated gastric juice,the same below.
2.2.2 芯壁比对包埋率的影响
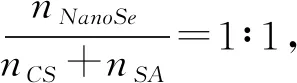
2.2.3 CaCl2用量对包埋率的影响
如图6(1)所示,改变交联剂CaCl2用量,采用“1.2.3.3”流程制备出的不同复合载药微球样品置于两种模拟消化液中1 h后,包埋率均出现急增缓减的变化趋势,最大包埋率对应的CaCl2用量为0.75 g(7.5 mg/mL)。实验现象说明交联剂CaCl2用量的增大并不能持续提高复合载药微球的药物包埋率,这是由于交联剂用量超过了饱和交联所需浓度时,多余的钙离子一部分游离于体系中使溶液的离子强度增大,剩余的部分钙离子会吸附在微球表面的高分子壁材壳膜上,导致其极性增强、亲水性增大[13]。两者结合致使复合载药微球样品的溶胀速度加快,药物包埋率降低。
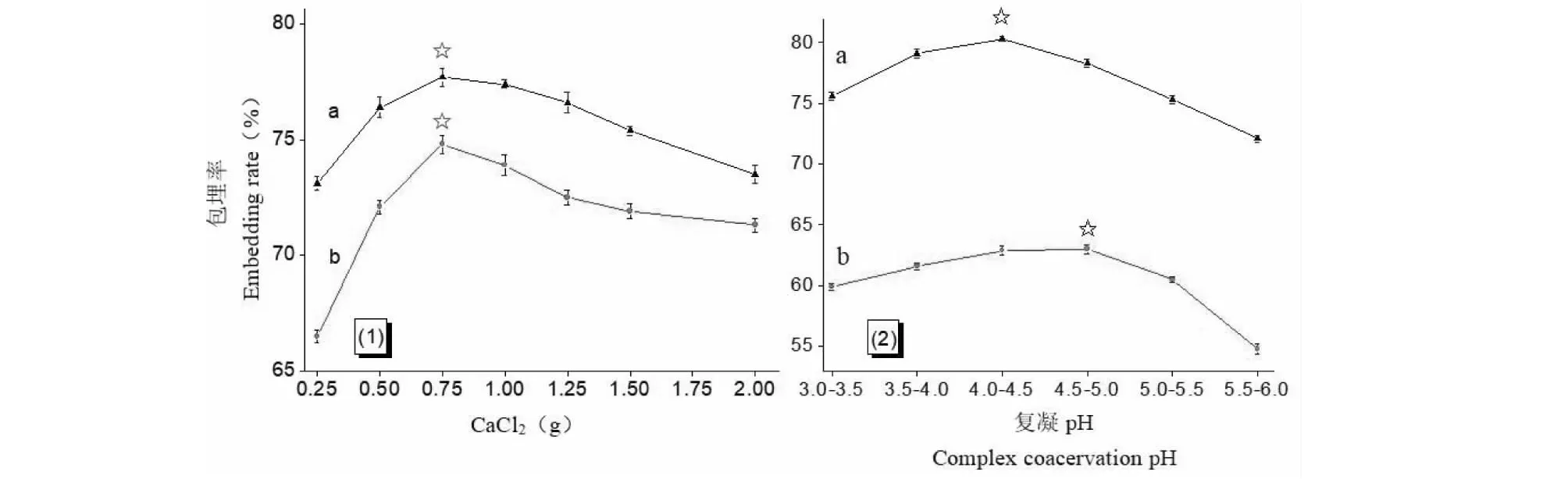
图6 CaCl2用量、复凝pH对载药微球样品包埋率影响Fig.6 The influence of dosage of CaCl2 and complex coacervation pH to the embedding rate of microspheres
2.2.4 复凝pH值对包埋率的影响
如图6(2)所示,随着复凝pH的增大,采用“1.2.3.4”流程制得的不同复合载药微球样品置于两种模拟消化液中1 h后,测得的微球包埋率均呈现先增后减的变化趋势。此种现象可能与海藻酸钠的自身性质有关,海藻酸盐沉淀pH值为3.6以下,溶解pH值为5.8[14],使用其作为壁材包裹芯材,当溶液酸性减弱时,有利于微球壁材中海藻酸钠溶解,导致微球壳膜加速崩解,药物释放率激增,包埋率骤减。图中,采用复凝pH=4.0~4.5制备的复合载药微球样品在模拟小肠液中具有最大的包埋率;而在模拟胃液中,虽然采用复凝pH=4.5~5.0制备的复合载药微球样品具有最大的包埋率,但与复凝pH=4.0~4.5时制备的微球样品的包埋率差别不大(△=0.1%)。综合考虑之下,为了减缓微球壳膜的溶解,提高包埋率,选取复凝pH=4.0~4.5作为制备条件。
2.3 响应面优化复合载药微球制备工艺与缓释性能分析
2.3.1 响应面实验设计与结果
在单因素实验结果上,根据Box-Benhnken设计原理构建响应面实验,因素及水平如表2所示。响应面试验设计及结果如表3所示,其中1~24号是析因实验,25~29号为中心试验,中心实验重复5次以衡量实验误差。
2.3.2 方差分析
使用Design-Exper8.0.6软件对表3中的实验数据进行多元回归分析,得回归方程:
包埋率=80.72+8.32A+5.47B+9.13C+4.10D-2.61AB-6.02AC+3.76AD-8.14BC-5.03BD+0.33CD-7.19A2-8.93B2-7.65C2-11.93D2(4)
由表4所示,该回归模型极显著(P<0.000 1),模型的相关系数R2=0.971 4,调整相关系数RAdj=0.942 7,失拟项P=0.070 9>0.05不显著,说明该回归模型(式4)能够充分拟合实验数据,可利用此回归模型优化复合载药微球的制备工艺。表3中:一次项A、B、C、D,交互项AC、BC、BD,二次项A2、B2、C2、D2极为显著;交互项AD显著;AB、CD不显著。比较各因素的F值,得出各因素对载药微球包埋率影响的主次顺序为:交联剂用量(C)>壁材比(A)>芯壁比(B)>复凝pH(D)。

表2 因素水平与编码Table 2 Factor levels and code
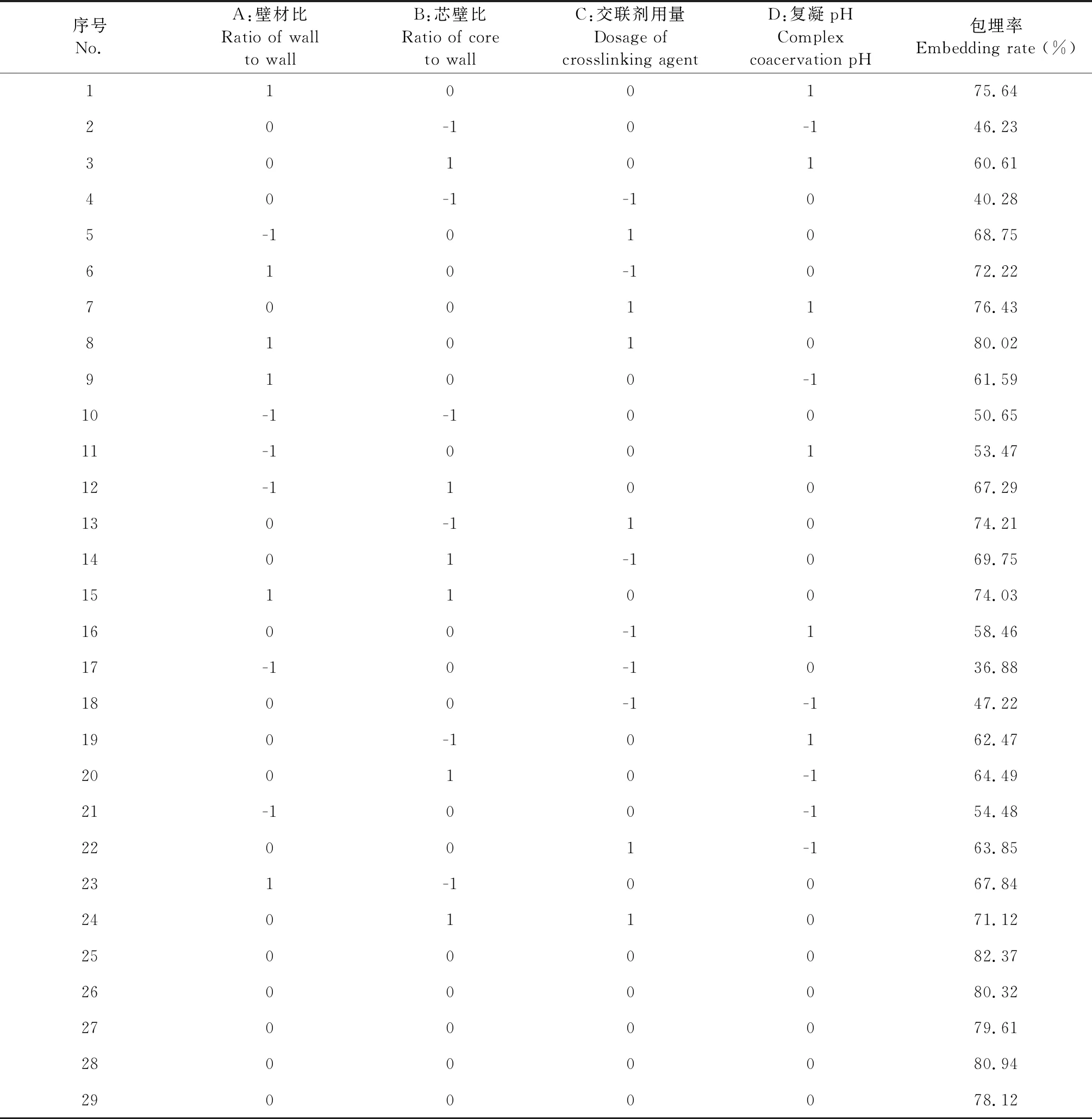
表3 BBD响应面设计方案与实验结果Table 3 Box-Behnken experimental design and results
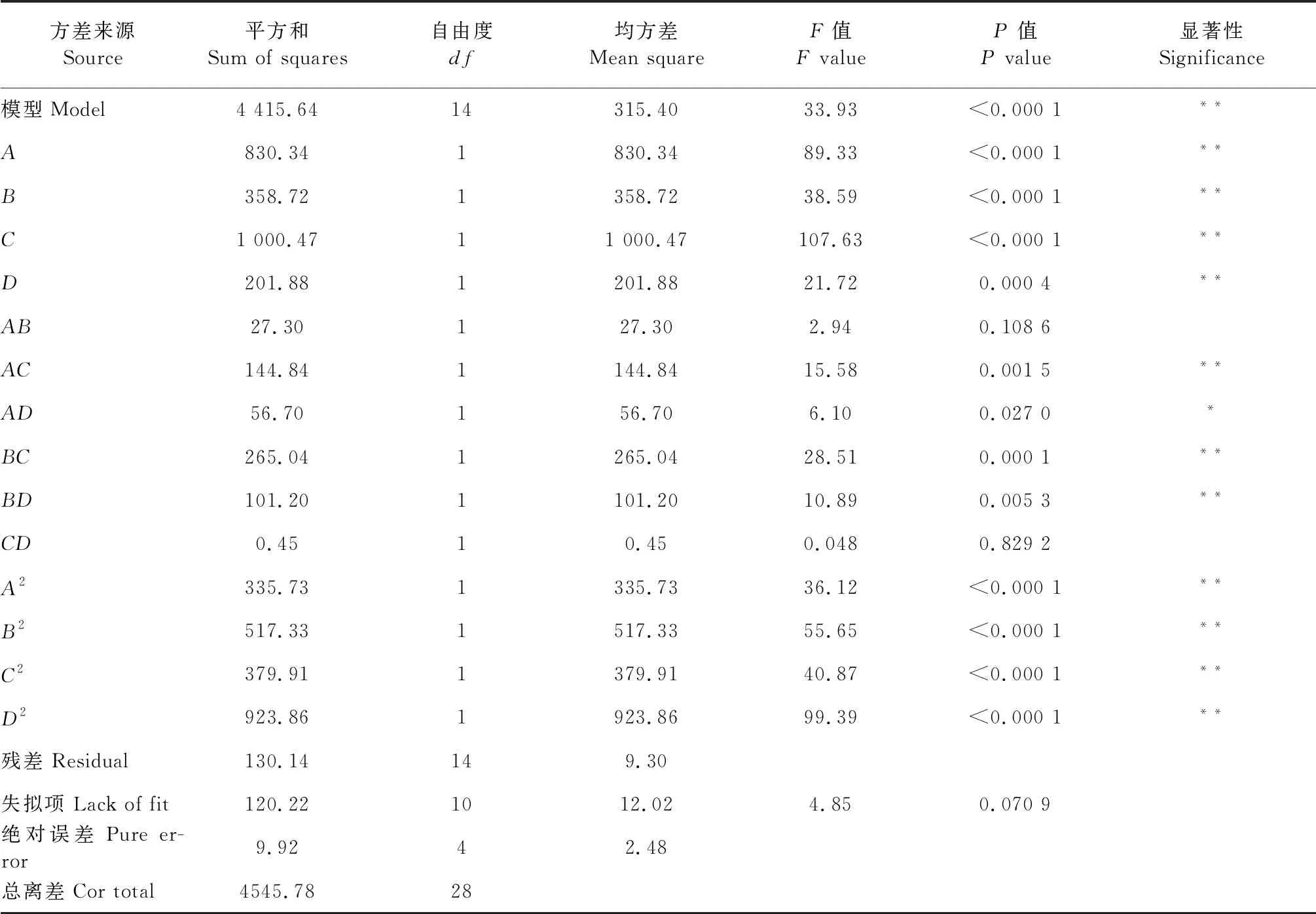
表4 回归方程的方差分析Table 4 Analysis of variance for the developed regression model
2.3.3 响应面图分析与优化
为了更直观地探讨上述因素之间的交互影响,固定任意两个因素,考察另两因素间的交互作用。利用Design-Exper软件绘制两因素交互作用的3D响应面图,如图7所示。
3D响应面曲面坡度的陡峭程度反映因素间交互作用的强弱;曲面的最高点与等高线图的圆心点对应,指向给定的因素水平下因素交互作用的极值。图7中,AC、BC的交互作用峰面明显比其他4个交互作用峰面陡峭,说明在整个6个交互作用中AC、BC的交互作用是最显著的,这与表3方差分析中AC、BC交互作用的P值(PAC=0.001 5、PBC=0.000 1)分列倒数第一和倒数第二相吻合。
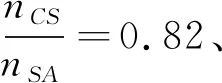
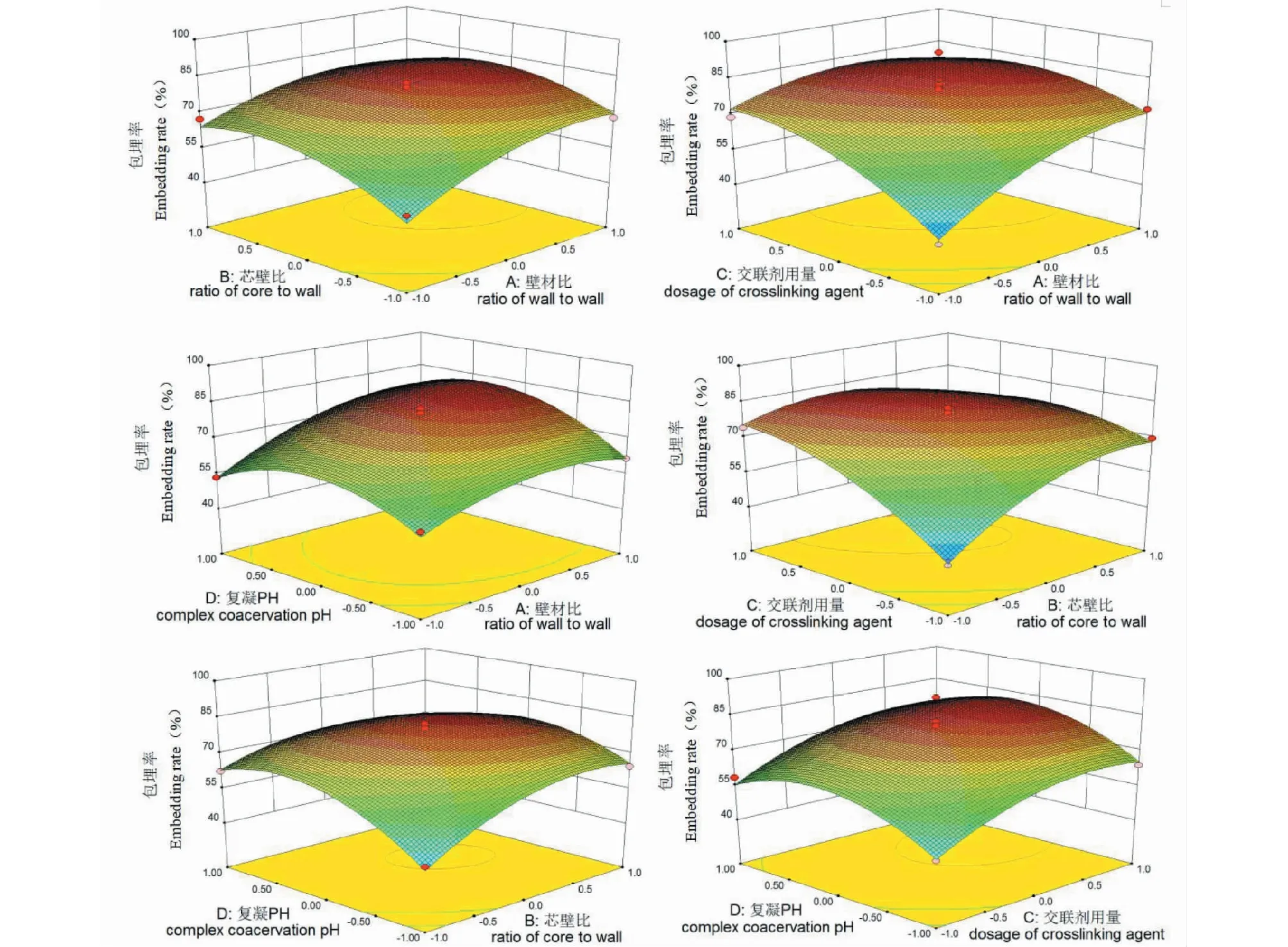
图7 两因素交互作用对包埋率影响的响应面图Fig.7 Response surface plots of embedding rate showing the interactive effects of two factors
2.3.4 体外模拟缓释性能验证
依据“1.2.3”体外释放实验方法,考察优化后的复合载药微球在两种模拟消化液浸泡(0.5~12 h)后的释放效果,如图8所示。
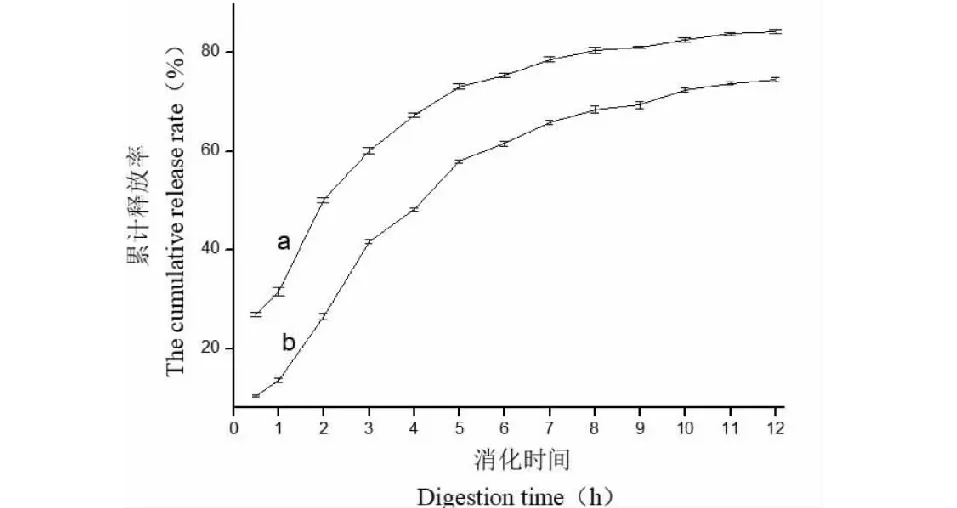
图8 优化后的复合载药微球体外模拟缓释性能Fig.8 The in vitro slow-release performance of optimized composite carrier drug microsphere注:a:模拟胃液;b:模拟小肠液。Note:a:Simulated gastric juice; b:Simulated small intestinal juice.
实验表明,优化后的复合载药微球在(0.5~12 h)时间范围内的药物累积释放率呈现“慢―快―慢”的变化趋势。微球在模拟消化液中浸泡的初期,致密的高分子壁材壳膜还来不及全面溶胀崩解、其结构尚完整,只有微球表面粘附和浅表层夹带的少量NanoSe开始释放,对应第一阶段“慢”释放(图8中0.5~1 h);随着浸泡时间的延长,高分子壁材壳膜由表及里开始全面溶胀崩解,大量释放通道出现,内部芯材的药物释放速度加快,表现为一定程度的突释,对应第二阶段“快”释放(图8中1~5 h);随着微球壳膜的彻底破坏及芯材表层中药物浓度的降低,微球内部载药向外扩散的难度逐渐增大,药物扩散速度开始减慢,进入第三阶段“慢”释放(图8中5~12 h)[15,16]。
相同时间下,复合载药微球在模拟胃液中的累积释放率大于其在模拟小肠液中,说明消化液pH降低有利于药物释放。体外模拟消5、7 h,载药微球在模拟胃液和模拟小肠液中的累积释放率达到73.08%和57.95%、78.57%和65.80%。考虑到混合型食物消化排空进入大肠需要5~7 h,在此时间内,微球已将大部分药物平稳释放出去,缓控释效果达到预期设想。
3 结论
SEM形貌表征与激光粒径分析证实,以壳聚糖(CS)、海藻酸钠(SA)为壁材,纳米硒(NanoSe)为芯材,L-盐酸赖氨酸(LMH)作为复合营养物,采用一步复凝法制备的NanoSe-LMH-CS-SA复合载药微球具有较小的粒度分布,其表面不规则凸凹结构有助于增大微粒比表面积大,适合作为缓控释载体。红外光谱分析证实CS和SA通过静电作用复凝聚构成微球壳膜,复合营养物LMH在成球过程中与壁材存在一定的静电作用。
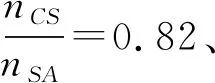
在体外模拟消化0.5~12 h范围内,载药微球的整个释药过程突释短、缓释长,体外模拟消化12 h,在模拟胃液和模拟小肠液中累积释放率分别达到84.29%和74.45%,证实本实验制备的NanoSe-LMH-CS-SA复合载药微球具备良好的缓控释作用。相比于现有的补硒制剂,微球负载的NanoSe具有突出的低毒高效性,同时复合的LMH可有效调节我国居民膳食性赖氨酸摄入不足,契合当前营养强化剂强调“功能复合”的设计开发思路,有望成为一种高效的复合缓释型补硒制剂。
