基于密度泛函理论的外电场下C5F10O的结构及其激发特性*
2020-01-16李亚莎孙林翔周筱陈凯汪辉耀
李亚莎 孙林翔 周筱 陈凯 汪辉耀
(三峡大学电气与新能源学院, 宜昌 443002)
采用密度泛函(DFT)B3LYP/6-311g(d)对C5F10O分子进行几何结构优化, 研究外加电场(0-0.03 a.u.,1 a.u.= 5.142 × 1011 V/m)对分子的几何结构、能量、前线轨道能级、红外光谱的影响.在相同基组下, 采用TD-DFT方法计算和分析C5F10O的轨道成分和激发特性.研究表明: 随着电场增加, 5C—15F与4C=16O键能逐渐减小, 键长增大; 13F原子的电荷布居数变化最快, 更容易在外电场力的作用下失去电子; 分子体系势能不断增加, 稳定性逐渐减低; 能隙EG值不断减小, 分子更容易激发到激发态参与到化学反应中.红外光谱中, 4个吸收峰发生蓝移, 4个吸收峰发生了红移.使用空穴-电子分析法, 指认了C5F10O分子前8个单重激发态的激发特征.第一激发态的激发能微小增长, 波长减小, 出现蓝移; 其余激发态的激发能均降低, 波长均变长, 发生红移, 导致C5F10O分子中的电子变得越来越容易激发, 体系的稳定程度减小.
1 引 言
六氟化硫(SF6)气体具有较高的绝缘性能和灭弧能力, 并且很难参与化学反应, 因此被广泛应用于大量高压设备中, 如气体绝缘封闭组合电器(GIS)、充气柜(C-GIS)、气体绝缘断路器(GCB)和气体绝缘管道母线 (GIL)等[1−4].然而,SF6气体的温室效应值 (GWP)约为 CO2的23900倍, 并且由于SF6的化学性质极为稳定, 能在空气中稳定存在3000多年[5,6].因此在1997年联合国气候变化公约缔约方签订了《京都议定书》, 将SF6列为限制性使用的温室气体之一, 限制其使用量[7].而使用N2、CO2气体与SF6混合,虽可以降低其使用量, 但通常需要进行高压压缩.考虑经济效益以及可行性问题, 寻找同SF6气体绝缘性能相当, 温室效应指数较低的新型环保型绝缘气体成为国内外研究的热点[8].
全氟五碳酮(C5F10O)不可燃、不破坏臭氧层、具有超低 (GWP ≈ 1)的全球变暖潜能值, 在大气中存在时间约为16 d, 并且电气绝缘强度为SF6的2.1倍, 可在多种绝缘设备中代替SF6, 是近年来发现的最有潜力替代六氟化硫的绝缘气体之一[9].瑞士ABB公司将C5F10O与空气混合, 用于22 kV开关柜并通过了根据IEC 62271-200-2001进行的应用性能测试[10].
目前针对C5F10O气体的研究主要有:C5F10O纯气和混合气体的介电性能研究[11−15],C5F10O与空气混合气体的有效电离系数研究[16],C5F10O/CO2混合气体的绝缘性能及其应用[17],C5F10O/CO2混合气体燃弧特性的研究[18],C5F10O/CO2和C5F10O/N2电弧等离子体热动属性与输运参数对比分析[19], 环保型绝缘气体C5F10O的构象分析[20], 氧气对C5F10O/N2/O2混合气体工频击穿电压和分解特性的影响研究[21],300—3500 K下C5F10O分解组分的变化特征[22].随着分子模拟技术的发展, C5F10O气体越来越多地应用于高电压绝缘领域[23].而外电场对C5F10O的分子结构及激发特性的影响研究还鲜有报道.
分子结构决定分子的性质, 为研究气体分子C5F10O的放电特性, 需要计算C5F10O的分子结构.在外电场的作用下, 气体中的少量电子加速撞击气体分子电离出更多的电子, 进而形成电子崩,因此分子在外场作用下的特性研究是许多领域的重要基础性工作.同时, 从长间隙气体放电的流注放电理论可知, 流注放电的发展和电离电荷引起的局部电场畸变以及光电离有关, 而光子的产生与气体的激发特性有关, 研究气体分子的激发特性有着重要意义.
本文利用密度泛函(DFT)B3LYP/6-311g(d)对C5F10O分子进行几何结构优化, 研究外加电场(0—0.03 a.u., 1 a.u.= 5.142 × 1011V/m)对分子的几何结构、能量、前线轨道能级、红外光谱的影响.然后在相同基组下, 采用TD-DFT方法计算和分析C5F10O的轨道成分和激发特征.所有的计算均在Gaussian 09软件包进行[24].考虑到气体分子之间较远的间距, 混合气体中各组分在电场中的特性应与纯气一致[25], 因此获得的结论亦可用于混合气体组成的系统, 并可为相关实验和装备的设计或进一步的理论研究提供参考.
2 理论计算
外电场作用下C5F10O分子的哈密顿量H可以用(1)式来表示[26]:

其中,H0为分子在未加电场时的哈密量,Hint为外电场与分子体系相互作用时的哈密顿量.在偶极近似的情况下, 分子体系与外电场相互作用能为

其中,µ为电偶极矩,F为外电场作用力.分子的动能和势能可由其算符通过分子波函数计算[27], 而分子波函数可以表示成单电子波函数的线性组合.因此, 动能算符的矩阵元表示为

核与电子间的吸引势能矩阵元表示为

电子与电子间的排斥势能是双电子积分, 其矩阵元表示为

(3)式—(5)式中的Dirac符号表示单电子的波函数.
根据Grozema等提出的模型[28,29], 在电场作用下的激发能Eexc与外电场作用力F, 电偶极矩和二阶极化率张量的变化量Dµ和Da满足关系:

公式中的冒号表示双重缩并.Eexc(0)为无电场下的激发能, 振子强度flu为[30]

其中, 线强度S为原子单位(),gl为加权因子,这里等于1,s表示波数.
3 计算结果及分析
3.1 外电场对C5F10O分子结构的影响
通过计算, 在能量最低时, 得到稳定的C5F10O基态分子结构模型, 如图1所示.在无外电场时, C5F10O分子键长与文献值[31]的对比如表1所示.
沿y轴负半轴方向加入外电场后(0—0.03 a.u.,1 a.u.= 5.142 × 1011V/m), 得到 C5F10O 分子键长和键角随电场的变化如图2、图3所示.4C=16O键 长 从 0.119086 nm缓 慢 增 长 为0.120626 nm, 5C—15F 键长从 0.132933 nm 增大为 0.136058 nm.这是由于 4C=16O键是由 2个sp2杂化轨道组成的双键, 原子间距离近, 键能较强, 不易在电场作用下发生变化; 而5C—15F键更长, 键能更小, 容易在外电场的作用下发生明显变化.同时由于分子电荷布居数的改变, 影响了各个原子之间的局部电场, 增大或减小了原子之间的键长[32].在这里, C=O和C—F键都是极性共价键,Mulliken电荷布居计算误差较大, 因此使用NBO 电荷布居数[33]计算, 如图4 所示.O、F 原子电负性强于C原子, 因此O、F原子整体带负电,C原子带正电.在电场力的作用下, 5C—15F与4C=16O之间的电荷布居所产生的吸引力逐渐增大, 使其在外电场力和原子作用力的合力下, 键能逐渐减小, 键长增大, 化学键更容易断裂.

表1 C5F10O 分子键长与文献值的对比Table 1.The bond length of C5F10O compared with the reference.

图1 C5F10O 分子的基态结构Fig.1.Stable structure of C5F10O.

图2 不同电场强度下 C5F10O 分子的键长变化Fig.2.Bond length of C5F10O at different electric field.
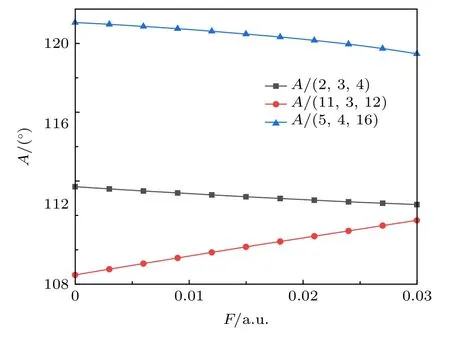
图3 不同电场强度下 C5F10O 分子的键角变化Fig.3.Bond angle of C5F10O at different electric field.

图4 不同电场强度下C5F10O的NBO电荷布局数Fig.4.NBO charge of C5F10O at different electric field.

图5 不同电场强度下 C5F10O 的电子云Fig.5.Electron cloud of C5F10O at different electric field.
根据图5, 我们可以清晰地看出C5F10O的电子朝着y轴正半轴移动, 这是因为电子逆电场运动, 16O原子周围的负电荷增多, 电荷布居数减小引起的.而位于同一垂直线上的6F原子和13F原子的变化速度不同, 位于上方的13F原子电荷布居数变化更快, 更容易在外电场力的作用下失去电子.
3.2 外电场对C5F10O分子能量的影响
根据计算结果, 得到不同电场下C5F10O分子总能量、动能以及势能的变化数据.由图6可知,随着外电场的增大, 分子体系的总能量E和动能Ek逐渐减小.这是由于正负电荷中心的不断偏移,以及分子极性的不断增大所导致的; 而分子势能则不断增大, 分子体系的稳定性是与势能相关的, 体系势能变大, 分子稳定性降低.

图6 能量随电场强度的变化 (a), (b), (c)分别是总能量、动能和势能随电场强度的变化Fig.6.Variation of energy of C5F10O at different electric field.Panels (a), (b), and (c) are changes of total energy,kinetic energy, and potential energy at different electric field.
3.3 外电场对C5F10O前线轨道的影响
采用相同泛函和基组, 得到C5F10O最高已占据轨道 (HOMO)能级EH和最低未占据轨道(LUMO)能级EL的数据, 由

可以得到分子能隙EG的数据.图7表示了分子处于基态时, C5F10O的前线轨道图.可以发现HOMO(MO 64)轨道主要由O原子孤对电子n轨道和3C—4C—5C的s轨道组成, 最容易被激发至LUMO(MO 65)轨道C=O键的π*空轨道中.

图7 C5F10O 的前线轨道图Fig.7.Molecular frontier orbital of C5F10O.

表2 不同电场强度下C5F10O的前线轨道能级Table 2.Frontier orbital energy levels of C5F10O at different electric field.
由表2可知, 随着外加电场的增大, LUMO能级逐渐减小, 电子更容易被跃迁到该空轨道中, 分子的亲电性增加; HOMO能级同样减小, 位于其能级上的电子更加稳定.能隙EG表示了电子从HOMO轨道跃迁至LUMO轨道的能力, 表现了分子活化参与化学反应的能力[34].从图8可知,EG值逐渐减小, 电子从HOMO轨道跃迁到LUMO轨道所需要的能量降低, 分子容易激发到激发态而变得更加活跃, 体系的稳定程度也就越小, 参与化学反应的能力增强, C5F10O绝缘气体老化程度也就随之增加.

图8 不同电场强度下 C5F10O 的能隙变化Fig.8.Energy gap of C5F10O at different electric field.
3.4 外电场对C5F10O红外光谱的影响
使用与优化相同的方法对C5F10O的振动频率进行了计算, 得到了在外电场为 0, 0.015, 0.030 a.u.时的数据, 如图9所示.

图9 不同电场强度下 C5F10O 的红外光谱Fig.9.Infrared spectrum of C5F10O at different electric field.
分子处于基态时, 记录其8个主要的吸收峰.通过对比可以发现, 位于 705.42 cm–1、1232.72 cm–1和1285.57 cm–1处的吸收峰对应1C—2C—3C—4C—5C的面内摇摆振动、面外摇摆振动和剪式振动, 均出现蓝移现象.这是由于在外电场的作用下, 改变了分子的电荷布居数, 使碳链整体键能增大, 红外光谱发生蓝移.位于 827.24 cm–1以及 1872.02 cm–1处的吸收峰分别归属于O=C—CC基团的不对称变形振动和4C=16O键的伸缩振动, 由于4C=16O键长增大, 导致键能减小, 吸收峰发生红移.位于 1183.46 cm–1、1210.73 cm–1处的吸收峰分别对应于以2C为中心的基团和以5C为中心的基团的对称变形振动, 出现红移现象, 键能逐渐变小,基团稳定性下降.位于 1333.81 cm–1处的吸收峰归属于1C—2C—3C的不对称伸缩振动, 出现蓝移现象, 其键能在外电场作用下不断增大, 基团更加稳定.
3.5 外电场对C5F10O激发态的影响
在上文C5F10O分子基态结构的基础上, 采用WB97XD/6-311g(d)方法计算得到C5F10O分子在无外电场下的激发数据.运用空穴-电子分析法考察分子的电子激发特征, 可以定量考察电子转移距离、空穴与电子的分离程度、分子轨道对空穴和电子的贡献程度、空穴与电子之间的激子束缚能(exciton binding energy).定义衡量空穴和电子质心之间距离的D指数、衡量空穴和电子的分离程度的t指数和激子束缚能EC[35]:

其中:Xele,Yele,Zele是电子质心坐标;Xhole,Yhole,Zhole为空穴质心坐标;HCT为空穴-电子的平均延展程度;rhole,rele为空穴分布和电子分布.在电子跃迁过程中, 被激发到激发态的电子和基态中残留的空穴由于库仑力相互作用, 将形成一个束缚态,称为激子, 其之间的空穴-电子库伦相互作用力, 则为激子束缚能.激子束缚能大, 表明空穴-电子之间的距离更近, 自由激子容易和杂质结合形成发光中心[36].根据计算, 可以得到无外电场下C5F10O分子前8个单重激发态的激发特性, 如表3所示.
使用Multiwfn可以绘制出C5F10O分子轨道跃迁图[35,37], 如图10所示, 其等值面为0.02结合图10和表3, 可以分析分子前8个单重激发态的激发特性.S(0)→S(1)中空穴和电子轨道分别主要由 MO 64 (HOMO)、MO 65 (LUMO)组成,D指数小,t指数明显为负, 空穴和电子分布之间没有显著的分离, 结合上图可以准确地判断为O原子n轨道 → C=O键π*轨道的局域激发, 印证了上文对前线轨道的激发预测.S(0) → S(2)空穴和电子 轨 道 主 要 由 MO 63 (HOMO-1)、 MO 65(LUMO)组成, 可以发现,D指数较大,t指数为正, 并且激子束缚能较小, 空穴-电子距离远, 应为2C—3C键的s轨道→C=O键π*轨道的电荷转移激发.

表3 C5F10O 前 8 个单重激发态的激发特性Table 3.Excitation characteristics of first 8 singlet-excited states of C5F10O.
S(0) → S(3)、S(0) → S(4)的激发数据相似:D指数不大,t指数为负数, 同时激子束缚能较大,为典型的局域激发.S(0) → S(3)对应 5C—4C=16O键的s轨道 → C=O键π*轨道的局域激发;S(0) → S(4)对应 C=O键 s轨道 → C=O键π*局域激发; S(0) → S(5)激发数据虽然与前两个激发态相似, 但将等值面调至0.01后, 根据图11,可以发现其应为O原子n轨道 → 碳链π*轨道的电荷转移激发.

图10 C5F10O前8个单重激发态的电子跃迁图(等值面为0.02)Fig.10.Electron transition of first 8 single-excited states of C5F10O (value = 0.02).

图11 C5F10O 第 5 个单重激发态的电子跃迁图 (等值面为0.01)Fig.11.Electron transition of the 5 single-excited states of C5F10O (value = 0.01).
S(0) → S(6)、S(0) → S(7)的激发数据同样相似:D指数大,t指数为正, 激子束缚能小, 可以认定为电荷转移激发.S(0) → S(6)为 13F、14F、15F原子孤对电子n轨道 → C=O键π*轨道的电荷转移激发; S(0) → S(7)为 10F、11F、12F 原子的n轨道→C=O键 π*轨道的电荷转移激发.S(0)→S(8)虽t指数略微负值, 但激子束缚能较小,并且从图10我们可以看出, 应为 9F、12F、13F、15F原子的n轨道 → C=O键π*轨道的电荷转移激发.
为进一步研究外电场对C5F10O分子激发态的影响, 计算了外电场下前8个激发态的激发能Eex、波长l和振子强度f, 计算结果如表4, 5, 6所示.
从表4, 5, 6 可以看出, 在 外电场作用下,C5F10O分子前8个单重激发态始终无禁阻跃迁,振子强度均不为0, 都能发生电子跃迁.其中第5激发态的振子强度最高为0.0591, 吸光率最强.结合前文对前8个激发态的指认可以看出, 电荷转移激发 (n= 6, 7, 8)所需要的能量普遍大于局域激发 (n= 1, 3, 4)所需能量, 只有第 2 激发态由于电子转移距离较短, 激发能较低.第 1激发态(n→ π*)的电子跃迁所需能量最小, 激发能为4.041 eV, 相应的吸收光波长为 306.80 nm, 在近紫外区.其余7个激发态的跃迁轨道能级较高, 吸收光波长在远紫外区.沿着y轴负半轴不断增加电场后, 除了第1激发态的激发能微小增长以外, 其余7个激发态的所需要的能量均降低.这也反应了除第1激发态的波长减小, 出现蓝移以外, 其余7个激发态的波长均变长, 发生红移, 导致C5F10O分子中的电子变得越来越容易激发, 体系的稳定程度也就越小.

表4 不同电场强度下C5F10O前8个单重激发态的激发能Table 4.Excitation energy of first 8 singlet-excited states of C5F10O at different electric field.

表5 不同电场强度下C5F10O前8个单重激发态的波长Table 5.Wavelength of first 8 singlet-excited states of C5F10O at different electric field.

表6 不同电场强度下C5F10O前8个单重激发态的振子强度Table 6.Oscillator strength of first 8 singlet-excited states of C5F10O at different electric field.
4 结 论
本文运用B3LYP/6-311g(d)方法对C5F10O分子进行了结构优化, 计算出不同外电场下对分子几何结构、能量、前线轨道能级、红外光谱的影响,同时使用含时密度泛函(TD-DFT)WB97XD/6-311g(d)方法对激发态和轨道成分进行研究和分析, 研究表明, 随着外电场强度沿着Y轴负半轴不断增加:
1) 5C—15F与4C=16O在电荷布居数的影响下, 键能逐渐减小, 键长增大.13F 原子的电荷布居数变化更快, 更容易在外电场力的作用下失去电子.
2) 分子总能量和动能逐渐降低; 分子体系的势能不断增加, 分子的稳定性逐渐降低.
3) LUMO能级和HOMO能级逐渐减小, 分子的亲电性增加; 能隙EG值不断减小, 分子更容易激发到激发态而参与到化学反应之中.
4) 红外光谱中, 随着外电场的增加, 位于705.42 cm–1、 1232.72 cm–1、 1285.57 cm–1以 及1333.81 cm–1处的吸收峰发生蓝移, 代表着振动所需的能量变高, 对应基团的键能增大, 基团更加稳定 ; 827.24 cm–1、 1872.02 cm–1、 1183.46 cm–1和1210.73 cm–1处的吸收峰发生了红移, 代表着振动所需的能量变低, 对应基团的键能减小, 基团更加不稳定.
5) 使用空穴-电子分析法, 考察了分子的电子激发数据, 指认了C5F10O分子前8个单重激发态的激发特征.第1激发态的激发能微小增长, 波长减小, 出现蓝移; 其余激发态的激发能均降低, 激发态的波长均变长, 发生红移, 导致C5F10O分子中的电子变得越来越容易激发, 体系的稳定程度减小.
