重芳烃加氢机理及热力学研究进展
2020-01-15范景新苏文利郭春垒孙磊马明超李健赵训志臧甲忠于海斌
范景新,苏文利,郭春垒,孙磊,马明超,李健,赵训志,臧甲忠,于海斌
(1中海油天津化工研究设计院有限公司,天津300131;2天津市炼化催化技术工程中心,天津300131;3山东滨化滨阳燃化有限公司,山东滨州256600)
随着车用柴油国Ⅵ标准在全国范围内陆续实施,要求柴油中稠环芳烃质量分数由11%降至7%,以2017 年柴油消费量1.68 亿吨计,新规的实施将导致从柴油池中挤出672万吨芳烃,通过柴油吸附分离可有效应对芳烃溢出效应,但吸附出来的富含稠环芳烃的重芳烃也需要拓展利用途径。
由于重芳烃富含芳烃的特性,通过加氢的方式促进其转化是实现重芳烃提质增效的有效途径,最大化转化重芳烃和生产高附加值轻质芳烃成为重芳烃转化的主要目标[3-5]。本文主要介绍了重芳烃加氢转化过程的反应机理和热力学研究进展,并对其发展趋势进行了展望。
1 重芳烃性质
2 加氢转化反应网络及机理
2.1 茚类和茚满的加氢转化
茚类被加氢饱和为茚满类后,再进一步发生加氢开环反应,从而得到理想的芳烃组分。Nylén等[8-9]以等体积浸渍法制备了10余种Pt-Ir 开环催化剂,发现Ir的相对负载量和载体的选择是影响催化剂反应性能、初始和最终产物分布的关键因素。由于Ir具有较高的初始氢解活性,能够减少积炭前体的生成,因此,增加Ir的负载量可以减少积炭,提高活性。Nylén等还根据茚满在Pt-Ir催化剂上反应的实验结果(325℃,常压),提出了茚满的开环反应机理,如图1所示。茚满的饱和环如果只发生一次断键,那么生成的产物主要为2-乙基甲苯和正丙基苯。由于一系列的脱烷基和氢转移作用,导致乙苯、邻二甲苯、甲苯、苯和C1~C6轻组分的形成。
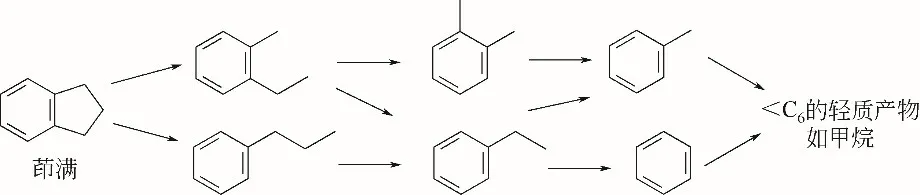
图1 茚满在Pt-Ir双功能催化剂上常压下的反应机理[8]
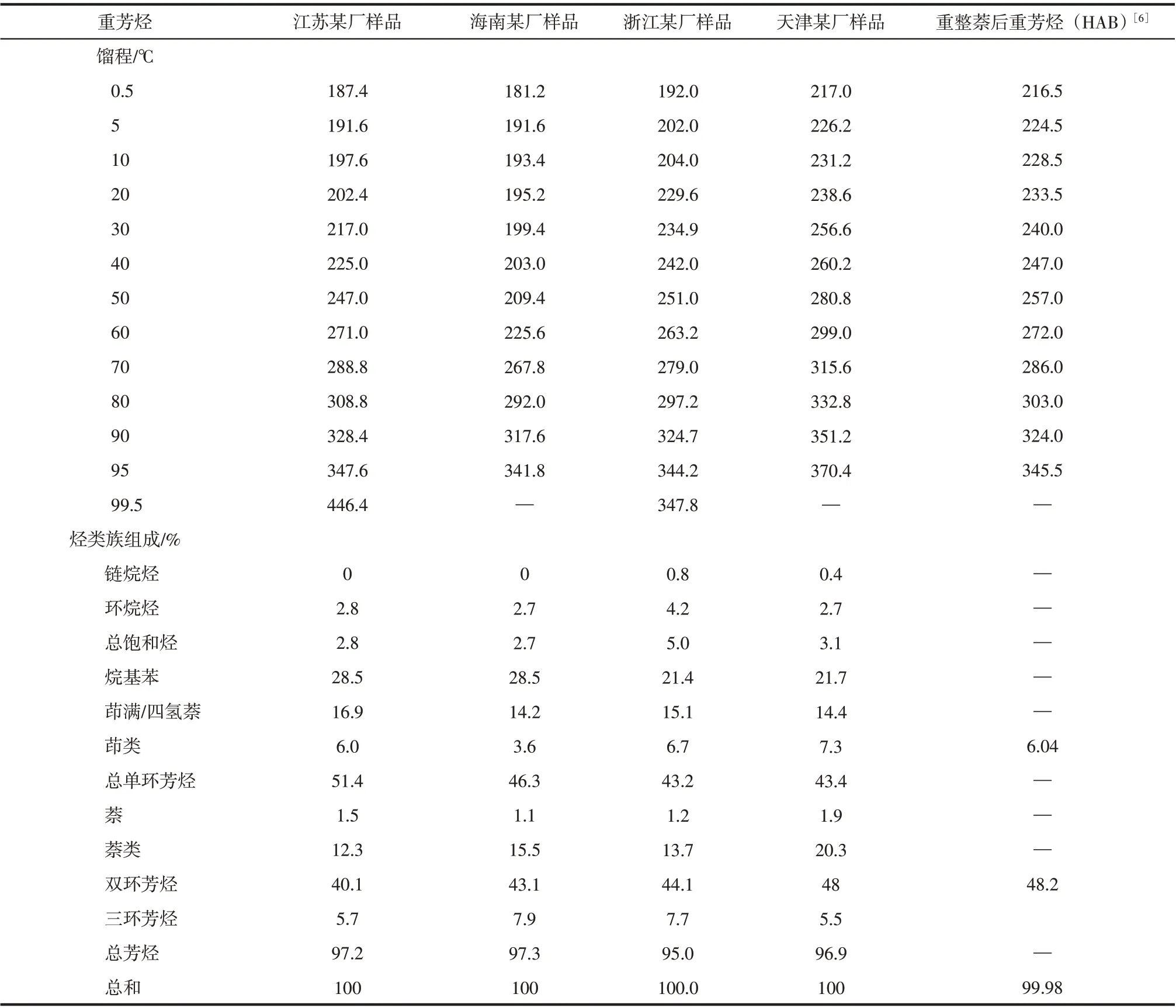
表1 代表性重芳烃性质
2.2 四氢萘的加氢转化
陈俊森[11]将双功能催化剂上四氢萘的加氢裂化反应网络进行了归纳,主要包括加氢、开环、脱氢、异构等12个反应,见图3。四氢萘的开环反应公认在催化剂的B酸位点发生,而由于金属表面溢流氢的存在,四氢萘的加氢反应不仅发生在催化剂的金属位上,也发生在酸性位上。随着金属位和酸性位点间距的增大,异构和开环反应速率减小。随着金属负载量的增大,异构和开环反应速率增大。此外,开环产物的收率和产品分布还与沸石形态结构有关,较强的酸性有利于开环反应[12-14]。
2.3 萘类的加氢转化

图2 模拟四氢萘热裂解机理模型中的典型反应(350~500℃)

图3 NiMo/HY催化剂上四氢萘加氢裂化反应网络
双功能催化剂上,萘与四氢萘的加氢转化路径及产物分布类似,简化的加氢转化反应路径见图4。首先芳环加氢饱和得到四氢化萘,然后再进行加氢裂化或者发生过度加氢反应。当反应温度较低时,萘主要发生加氢反应生成四氢萘。
由于萘分子在金属位上较强的吸附作用,萘加氢生成四氢萘的反应速率较四氢萘加氢生成十氢萘的速率快一个数量级[16]。与之相似,甲基萘加氢生成甲基四氢萘的速率比甲基四氢萘加氢生成甲基十氢萘的反应快一个数量级。同时,由于甲基的空间位阻效应等因素,2-甲基萘的转化速率比萘慢1.5倍[17-18]。因此,加氢反应速率大小顺序为萘>甲基萘>四氢萘>甲基四氢萘。
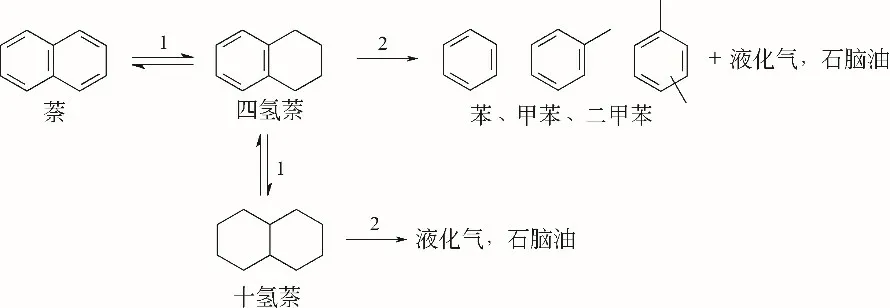
图4 萘的加氢裂化反应路径[15]
2.4 菲的加氢转化
多环芳烃要生成BTX,首先需要将多环芳烃部分芳环饱和,然后再进行加氢裂化反应。2-甲基菲的加氢饱和反应网络如图5所示,其加氢饱和反应过程中的主要物质包括[19]2-甲基菲(A)、氢气(B)、2-甲基-9,10-二氢菲(C)、2-甲基-1,2,3,4-四氢菲(D)、2-甲基-1,8-八氢菲(E)、2-甲基-1,10-八氢菲(F)、2-甲基全氢菲(G)、2-甲基-5,6,7,8-四氢菲(H)、2-甲基-5,10-八氢菲(I)。
菲首先加氢生成二氢菲,然后继续加氢生成四氢菲和八氢菲。在弱酸性NiMo/Al2O3催化剂上,二氢菲吸附在催化剂B酸中心,因为共轭作用形成的碳正离子更稳定,苯环上的二级碳优先发生断裂,从而形成2-乙基联苯;菲两端的饱和环断键遵循双功能机理,六元环异构为五元环,五元环再在酸性位上发生开环,这一过程主要生成丁基萘、甲基丙基萘和二乙基萘[20-21]。生成的萘类物质继续沿着图4所示萘类的加氢裂化反应路径进行反应,从而生成BTX 和低碳烷烃,BTX 的收率主要取决于各步骤的加氢选择性。菲的加氢裂化反应机理如图6。
2.5 蒽的加氢转化
蒽的加氢裂化同样要先经历芳环的加氢饱和,再发生饱和环开环断裂等反应。蒽分子加氢分为两种路径:中环加氢和端环加氢[22-23](图7),但两个路径所需消耗的氢气量区别很大。通过中环加氢再开环断裂,仅需消耗3个氢气就可以将1个蒽分子变成2 个低碳芳烃;如果从端环开始加氢开环反应,则需消耗7个分子的氢气,才能得到一个分子的汽油组分;如果从端环开始加氢反应,同时又存在过度加氢,则需消耗更多的氢气。因此,如果能够控制催化反应过程路径,不但能提高产品质量和目的产品选择性,亦可大幅降低氢耗和能耗。
实验研究表明,选择适当的催化剂和反应条件是可以实现促进蒽分子中环加氢反应[24]。比如,蒽在300℃下负载于镍的凹凸棒土(Ni/AP)上会发生选择性加氢生成二氢蒽,而在175℃下完全转化为全氢蒽,这是由于175℃下,Ni/AP 会有效促进双活性氢的形成和转化(H…H),从而导致蒽全加氢,而在300℃下,Ni/AP催化产生的H…H倾向于均裂成氢自由基(H˙),从而导致蒽部分加氢为二氢蒽。因此,活化氢物种是蒽催化加氢反应选择性的重要影响因素[25]。

图5 2-甲基菲的加氢反应网络
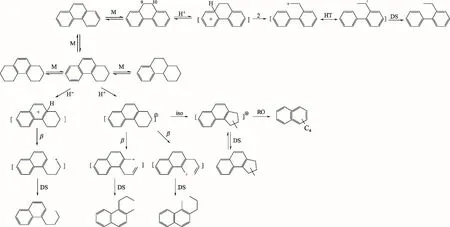
图6 菲的加氢裂化反应机理
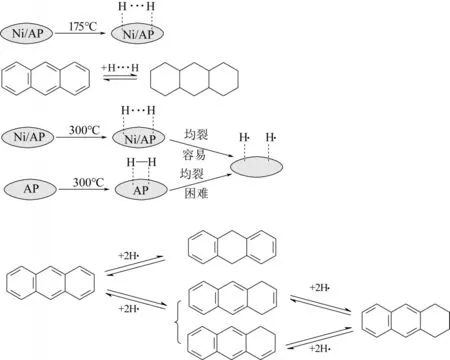
图7 不同温度条件下蒽在Ni/AP上的加氢反应
3 加氢裂化反应热力学
在多环芳烃加氢反应体系研究中,以反应路径和机理研究为基础,如果通过热力学研究的方式得到各反应平衡常数,那么动力学研究时需要估算的反应动力学参数数量就会大幅减少,并可以保证正逆反应速率常数的热力学一致性,所以加氢热力学分析对于反应动力学研究是必要的。同时,热力学研究也可以为优化重芳烃加氢反应条件提供参考。在众多热力学研究方法中,基于反应过程物质的分子结构,通过官能团贡献法估算分子热力学性质的方法[26-27]出现较早,受到普遍认可,可以通过热力学计算得到多环芳烃加氢反应体系中各反应在不同温度下的平衡常数、焓变、吉布斯自由能变、熵变等。
Contreras 等[28]同时运用密度泛函理论(DFT)与Benson 官能团贡献法计算了1-甲基萘加氢反应的热力学平衡常数,使用的反应路径模型简图见图8,平衡常数计算值见表2。两种方法计算的平衡常数数值虽有一定偏差,但都反映出1-甲基萘的第一个芳环加氢饱和平衡常数要大于第二个芳环加氢饱和平衡常数,且不含取代基的芳环比含有取代基的芳环更容易加氢。

图8 1-甲基萘的第一和第二个芳环加氢反应模型简图

表2 673K温度下计算的平衡常数
彭冲[18]在研究萘的加氢反应时得出了相似的结论:萘第一个环加氢的平衡常数较大,生成四氢萘后第二个环加氢的平衡常数较小。另外,萘第一个环加氢生成四氢萘后,异构和开环生成甲基茚满和丁基苯的平衡常数都很大。随着反应温度的升高,四氢萘加氢生成十氢萘的反应平衡常数下降很多,而四氢萘异构和开环反应的平衡常数变化不大,所以从热力学角度来讲,高温有利于异构和裂化反应,减少过度加氢,进而有利于增产BTX。
单就三环芳烃加氢饱和反应来讲,在蒽加氢体系中,随温度升高,部分加氢产物四氢蒽的平衡含量存在最大值,在热力学上蒽加氢生成trans-syntrans-式的全氢蒽比生成cis-trans-式的全氢蒽更易自发进行。在5MPa、350℃时,蒽接近完全转化;在菲加氢体系中,随温度的升高,部分加氢产物四氢菲的平衡含量也存在一个最大值,在热力学上trans-anti-trans-式的全氢菲比其他全氢菲更易自发生成[29]。
在双功能催化剂上,菲加氢生成二氢菲的反应平衡常数大于生成四氢菲的反应平衡常数,并且菲加氢生成四氢菲的反应平衡常数受温度变化影响更大,所以热力学上来讲,菲更倾向于加氢生成二氢菲。但由于二氢菲中间环的空间位阻效应,其开环转化效率较低,因此反应大部分沿着四氢菲路线进行。如果可以提高二氢菲中环开环反应的效率,就可以仅仅使用3 分子氢气获得2 分子的优质汽油组分[18]。
受热力学因素影响,不同环数和饱和程度的芳烃,其最优的加氢裂化反应条件有所区别:十氢萘在Pt-Ir/HY催化剂上的加氢裂化反应热力学平衡计算结果表明[30],由于受到反应熵变的影响,环烷环内部C C键比环烷环外部C C键和非环烷烃C C键更难发生断裂。提高温度有利于促进环内C C键的断裂反应。十氢萘的总开环产物产率随氢烃比的增加而增加,当氢烃比增加到4时,总开环产物产率达到最大值,继续增大氢烃比对总开环产物产物基本没有影响。陈俊森[11]对四氢萘加氢裂化反应的热力学研究结果表明,为了多产单环芳烃,四氢萘加氢裂化适宜的反应条件为:400~420℃、5MPa、氢烃摩尔比6[10]。与十氢萘加氢裂化反应相比,四氢萘加氢裂化反应氢烃摩尔比的最优值较大,主要是由于其反应路径需要消耗更多的氢。彭冲[18]也认为高温、低压适合二环芳烃、三环芳烃定向转化为单环芳烃,反应温度宜选择400~420℃,菲加氢裂化的最优氢烃摩尔比为萘最优氢烃摩尔比的一倍[18]。而芳烃加氢饱和是高放热反应,消耗1mol 氢气反应放出的热量为63~71kJ。因此,不同环数和饱和程度的芳烃,加氢过程的放热也存在较大区别。
在实际操作和工业生产中,需充分考虑重芳烃原料自身特点和目的产物,调整反应条件。较常规柴油加氢裂化而言,由于重芳烃富含芳烃的组成特点,反应过程放热相对较高。工业化过程中需要针对这一特点,控制单程转化率、加强反应器的设计(控制催化剂床层温度)或采用不同性能的催化剂进行复配,避免造成飞温。对于重芳烃加氢转化增产低碳芳烃来讲,适宜的反应温度为400~420℃,反应温度过低,不利于单环芳烃的保留,而反应温度过高,只能小幅度增加单环芳烃的收率,但氢耗和能耗会增加,催化剂也易结焦失活;降低压力有利于提高单环芳烃的平衡收率,但须保证足够的压力以维持催化剂的稳定性,稠环芳烃适宜的反应压力在5MPa 左右;最佳氢烃摩尔比随重芳烃原料中稠环芳烃含量和芳环数的增加而增大。另外,如果能精确控制重芳烃的催化反应过程路径,不但可以提高产品质量和目的产品选择性,亦可大幅度降低氢耗和能耗;相反,若要生产低芳烃含量的油品或溶剂,可以适当降低反应温度,提高反应压力和氢烃比。
