距离和疏水模型辅助的蛋白质结构预测方法
2020-01-14王小奇周晓根张贵军
王小奇,周晓根,胡 俊,张贵军
1(浙江工业大学 信息工程学院,杭州 310023)2(密歇根大学计算医学与生物信息学系,密歇根州安娜堡 45108,美国)
1 引 言
蛋白质是生命活动的物质基础,对人类的健康起着决定性作用.准确预测蛋白质的结构对疾病研究、药物设计等方面都有重要的科学意义[1].目前测定蛋白质结构的实验方法主要有X射线晶体衍射、多维核磁共振和冷冻电镜.但是这些实验测定的方法代价昂贵[2-4],同时对于药物设计的主要靶标—膜蛋白而言,实验方法极难测定其结构.因此,根据Anfinsen法则[5],从氨基酸序列出发,利用计算机技术设计适当的算法从头预测蛋白质结构已成为生物信息学研究中的热点[6].
从头预测方法近十年来迅速发展,取得了很大的进步,尤其是华盛顿大学Baker研究团队开发的Rosetta[7,8]和密歇根大学Zhang研究小组开发的QUARK[9,10]在最近几届CASP[11-13]比赛中表现突出,但针对一些序列较长的蛋白(序列长度大于100),现有的方法预测精度依然不高[11,14].这主要是因为现有方法的构象空间采样能力不足,搜索不到近天然态的构象,并且能量函数的不精确性导致每个构象难以量化,增加了该问题的复杂性,进而影响了从头预测精度.
针对构象优化问题,国内外研究者相继提出了大量的构象空间优化方法,如进化算法[15-18]、蒙特卡罗[19,20]、构象空间退火[21-24]、副本交换[25]等被广泛应用于蛋白质结构预测中.研究表明,蛋白质结构先验知识可以降低能量函数不精确带来的误差,并有效地减小构象的搜索空间,进而提高蛋白结构的预测精度[26-29].APL[30]利用二面角信息来提高蛋白质结构的预测精度;RMA[14]和AIMOES[31]分别利用预测的二级结构来增强构象空间的采样;EVfold[32]和PconsFold[33]从预测的蛋白质接触图(contact map)中提取结构信息,并应用于蛋白质结构预测中;CONFOLD[34]和SCDE[35]利用二级结构和接触图来预测蛋白结构.DeepMind开发的AlphaFold系统[36]在CASP13的表现非常突出,主要是因为通过神经网络预测氨基酸之间的距离和化学键角度,然后再根据两种物理属性对结构进行评分,最后通过梯度下降优化,这也进一步证实了先验的结构知识对预测蛋白质结构的有效性.
尽管接触图可以提高蛋白质结构的预测精度,但是传统的接触图仅仅估计了每对残基间的距离是否大于某一阈值(8Å).距离谱[9,10]可以反应残基间的距离分布信息,基于距离谱作者前期在进化算法的框架下[37-39],提出了一种蛋白质构象空间优化方法[40]表明距离谱可以有效地提高预测的精度.然而,在蛋白质的折叠过程中,疏水作用是主要作用之一,对蛋白质结构的形成与维持有着特殊的作用[41],也被广泛地用来预测蛋白质结构,但现阶段的研究中氨基酸的疏水性主要针对于简化的HP模型[42,43].
针对现有预测方法的不足,在进化算法的框架下提出一种距离和疏水模型辅助的蛋白质结构预测方法(DHMA).首先,根据亲疏水氨基酸的回转半径来指导构象采样;然后,利用距离谱构建距离分布模型和疏水概率模型来指导种群的更新,进而提高预测的精度.在10个测试蛋白的实验结果表明,DHMA是一种有效的蛋白质结构预测方法.
2 DHMA算法设计
在DHMA中,根据氨基酸的亲疏水性与距离谱,分别构建亲疏水性氨基酸的回转半径、距离分布模型和疏水概率模型来指导构象的优化.
2.1 亲疏水氨基酸的回转半径差
在变异操作中,利用亲疏水氨基酸的回转半径差来引导目标个体的变异.
疏水氨基酸的回转半径定义如公式(1):
(1)

同理,计算得到亲水性氨基酸的旋转半径RGP,并且根据如下公式计算亲疏水氨基酸的旋转半径差:
DRG=RGP-RGH
(2)
在稳定的蛋白质结构中,疏水氨基酸残隐藏在蛋白质分子内部.因此在变异操作中,首先判断片段组装后的新个体是否满足DRG>0,如果DRG>0,则再采用能量函数进行评估;否则直接拒绝此次片段插入.
2.2 距离分布模型
在选择过程中,首先利用基于距离谱的打分模型来评价构象的质量.距离分布打分模型计算如式(3):
(3)
其中U是距离谱中残基对的集合,p和q是U中的任一残基对,dpq为构象中第p个残基与第q个残基的Cα原子间的欧氏距离,Npq(dpq)是在残基对(p,q)的距离谱中dpq对应的片段数.
距离分布估计模型的分值在很多程度上可以反应构象的质量,即Ds值越大表明构象的结构越好.因此,在选择过程中,首先利用距离分布估计模型来指导种群的更新.
2.3 疏水概率模型
在稳定的蛋白质结构中,疏水氨基酸隐藏在蛋白质分子内部而彼此相互接触.在距离谱中,只统计了欧式距离小于9Å的残基对间的距离分布信息,因此利用距离谱中疏水残基对的疏水性设计了打分模型,具体定义如式(4):

(4)
其中f(p,q)计算如下:
(5)
其中cp和cq分别是距离谱中第p和q个残基的疏水值[44].
疏水概率模型pH根据如公式(6)计算:
(6)
其中Hs与Hs′分别代表目标个体与变异个体的疏水得分,其中T根据如下公式计算:

(7)
其中u(p,q)定义如下:
(8)
同理可以计算得到变异个体的T′值.
在选择操作中,当变异个体的距离分布值比目标个体低时,利用疏水概率模型来辅助指导种群的更新,进一步提高DHMA的预测精度.
2.4 算法描述
DHMA算法的流程描述如下:
Step 1.初始化:进行参数初始化和种群初始化,具体过程如下:
Step1.1设置种群规模NP、温度常数kT、拒绝升温常数m、片段长度frag、迭代常数λ∈(0,1)和最大进化代数Gmax;利用Rosetta的第一和第二阶段产生NP个个体作为初始种群Pinit={x1,…,xNP};
Step1.2设置目标个体的索引i=1,i∈[1,NP],进化代数g=1;
Step 2.变异操作:利用亲疏水氨基酸的回转半径差和能量函数指导目标个体xi的变异,具体过程如下:
Step2.1利用片段组装技术对目标个体xi进行片段插入,并根据公式(2)计算组装后个体的DRG;若DRG≤0,则拒绝此次片段插入,否则计算组装前后个体的能量变化,并根据蒙特卡罗(Monte Carlo)机制判断是否接收此次片段的插入;
鉴于东海2号机组于1978年投运,原子能电力已于2017年11月向规制委提交延寿申请。要想让这份申请获得批准,该公司必须在2018年11月底这一法定截止日之前获得规制委对下述两份计划的批准:安全强化措施详细执行计划和延寿改造详细计划。
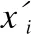
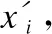
Step3.2若Ds>Ds′,则目标个体xi被保留,否则根据公式(6)计算疏水概率pΗ;

Step 4.判断是否结束:具体步骤如下:
Step4.1若i=NP,则设i=1,否则i=i+1,并且转至步骤Step 2;
Step4.2若g>λGmax,则frag=3,否则frag=9;
Step4.3若g≥Gmax,则结束迭代,否则g=g+1,且转至Step 2.
3 实验和结果
3.1 实验材料
在实验中,从PDB库中选取序列长度从64到125的10个测试蛋白,这些测试蛋白的折叠类型包括α、β和α/β,测试蛋白的具体信息如表1中的第2-4列所示.选取这些蛋白质做测试是因为它们在生物体内有着重要的作用[45,46],并且在现有文献中这些蛋白被广泛地研究[14,47,48],因此,这些蛋白可以较好地测试DHMA算法的性能.
测试蛋白的片段库和距离谱分别通过ROBETTA[49]和QUARK服务器获得,并且在构建片段库和距离谱的过程中均去除同源模板.实验的参数设置如下:种群规模NP=100、温度常数kT=2、拒绝升温常数m=150、迭代常数λ=0.6、最大迭代次数Gmax=1.5L,其中L为序列长度.在初始化过程中能量函数采用Rosetta的能量函数score0和score1,在变异操作中采用Rosetta的能量函数score3[7,8].通过计算预测模型与天然态结构中Cα原子的均方根偏差(RMSD)和TM-score[50]来评价算法的预测精度.本次实验中,独立运行DHMA算法10次,保留中间采样的构象,进而产生一个构象池,在同样的代价下,独立运行1.5L条蒙特卡罗轨迹并保留第三和第四阶段采样的所有构象.
3.2 结果分析
实验中DHMA的预测结果与Rosetta和QUARK预测的结果进行比较,不仅因为Rosetta和QUARK是目前最成功的预测方法之一,主要是因为DHMA算法使用了QUARK预测的距离谱,同时DHMA算法的初始阶段和变异过程中都采用了Rosetta的能量函数.表1和表2中的DH、Ro和QU分别是DHMA、Rosetta和QUARK的简称,其中QUARK的预测结果直接通过QUARK在线服务器直接得到.DHMA算法与Rosetta的预测结果主要分3步得到:
表1 第一个聚类结构的TM-core
Table 1 TM-core of first cluster structure
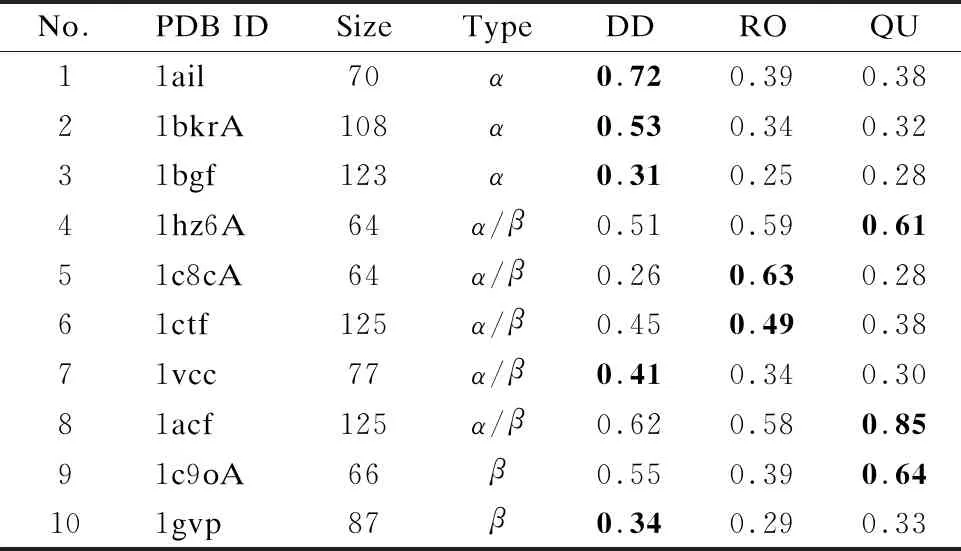
No.PDB IDSizeTypeDDROQU11ail70α0.720.390.3821bkrA108α0.530.340.3231bgf123α0.310.250.2841hz6A64α/β0.510.590.6151c8cA64α/β0.260.630.2861ctf125α/β0.450.490.3871vcc77α/β0.410.340.3081acf125α/β0.620.580.8591c9oA66β0.550.390.64101gvp87β0.340.290.33
1)在构象池中,根据能量函数选取能量最小的前10000个结构;
2)利用K-均值聚类算法进行聚类,选出聚类中心能量最小的一个类;
3)选出与类心最近的5个结构作为最后的预测结果.在聚类过程中,聚类半径为能量值,聚类簇数为5.
表1是DHMA、Rosetta和QUARK聚类得到的第一个结构与天然态结构比对的TM-score值.如表1所示,蛋白1ail、1bkrA、1bgf、1vcc和1gvp的结果表明DHMA的预测精度均优于Rosetta和QUARK,并且其中1ail和1bkrA的预测结果要明显优于Rosetta和QUARK.但对1acf和1c9oA蛋白来说,QUARK要明显优于DHMA和Rosetta;对1c8cA和1ctf蛋白来说,Rosetta的预测精度优于DHMA和QUARK.总体而言,DHMA的预测精度优于Rosetta和QUARK,是一种有效的蛋白结构预测方法.
表2 DHMA、Rosetta和QUARK的实验结果比较
Table 2 Comparison of results of DHMA,Rosetta and QUARK
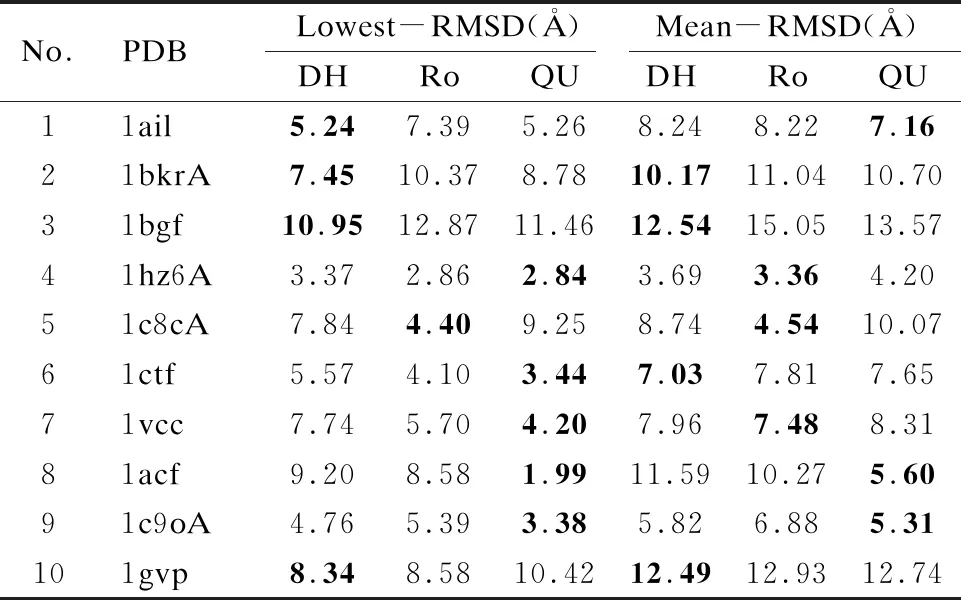
No.PDBLowest-RMSD(Å)Mean-RMSD(Å)DHRoQUDHRoQU11ail5.247.395.268.248.227.1621bkrA7.4510.378.7810.1711.0410.7031bgf10.9512.8711.4612.5415.0513.5741hz6A3.372.862.843.693.364.2051c8cA7.844.409.258.744.5410.0761ctf5.574.103.447.037.817.6571vcc7.745.704.207.967.488.3181acf9.208.581.9911.5910.275.6091c9oA4.765.393.385.826.885.31101gvp8.348.5810.4212.4912.9312.74
表2中Lowest-RMSD代表预测得到的最低RMSD值,Mean-RMSD代表预测得到的平均RMSD值.对于测试蛋白1ail、1bkrA、1bgf和1gvp来说,DHMA预测模型的Lowest-RMSD都优于Rosetta和QUARK,尤其是1bkrA和1c8cA,DHMA算法的Lowest-RMSD比QUARK和Rosetta的精度高于1Å.虽然,DHMA算法的Lowest-RMSD结果比QUARK预测的差,但是DHMA预测的Lowest-RMSD优于Rosetta预测的结果,其中对于1hz6A、1ctf和1vcc蛋白,DHMA预测的Lowest-RMSD比QUARK的差,但是在Mean-RMSD方面,DHMA算法的预测精度优于QUARK算白,DHMA预测的Lowest-RMSD比QUARK的差,但是在Mean-RMSD方面,DHMA算法的预测精度优于QUARK算法.同时,10个蛋白质中有4个蛋白,DHMA预测的Mean-RMSD优于QUARK和Rosetta预测的结果.总体而言,在Lowest-RMSD方面,DHMA比QUARK算法的预测结果差,但是在Mean-RMSD方面,DHMA算法的预测精度优于Rosetta和QUARK算法.
图1是DHMA、Rosetta和QUARK预测的结构与天然态结构之间的比对图.对蛋白1acf来说,DHMA和Rosetta结构在β区域均折叠错误,因此 QUARK的结构与天然态的结构更相似,但对1ail来说,DHMA得到的结构与天然态结构比Rosetta和QUARK的结构重叠区域更多,更相似.
3.3 采样能力分析
在采样能力分析中,本文将DHMA算法与Rosetta进行比较.部分蛋白的采样结果如图2所示,图中横坐标为构象与天然态结构之间的RMSD值;纵坐标为每个构象的能量值.

图1 DHMA、Rosetta和QUARK预测结构与天然态结构的比对Fig.1 Comparison between predicted structure by DHMA、Rosetta and QUARK and native structure
如图2所示,针对蛋白来1acf、 1bgf和1c9oA,DHMA算法增强了近天然态区域的采样,尤其对1bgf来说DHMA显著的增强了低RMSD区域的采样能力.但是对图2中的1hz6A蛋白来说,Rosetta比DHMA算法具有更好的采样能力,这主要是因为现有的能量包含足够多的信息,对一些序列较短的蛋白来说,现有的能量函数可以有效地指导构象的采样.
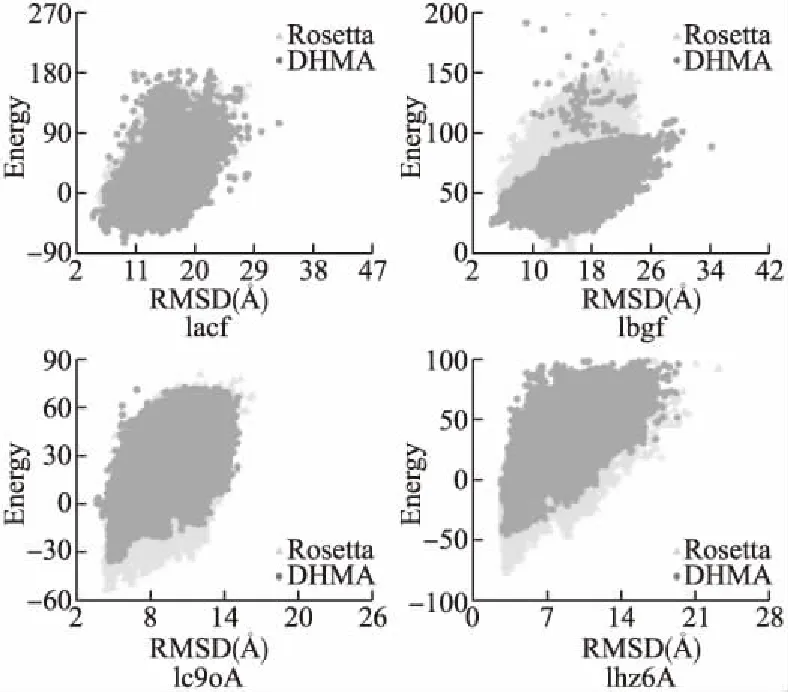
图2 构象空间采样比较Fig.2 Comparsion of conformation space sampling
4个蛋白的RMSD分布如图3所示,图中横坐标为构象与天然态结构之间的RMSD值;纵坐标为每个区域内构象数占所有构象总数的百分比.如图3所示,针对大多数蛋白来说,DHMA算法增强了低RMSD区域的采样.但对1hz6A蛋白来说,Rosetta比DHMA算法具有更好的RMSD分布,因此对1hz6A蛋白质来说,Rosetta的聚类结果比DHMA的精度高.
总体而言,DHMA算法能够在构象空间进行更充分的采样,采样得到蛋白质近天然态构象.这主要是DHMA算法采用了距离分布和疏水模型指导构象的采样,有效的弥补了能量函数不精确带来的误差.

图3 RMSD分布比较Fig.3 Comparison of RMSD distribution
4 结 论
本文提出一种距离和疏水模型辅助的蛋白质结构预测方法.在进化算法的框架下,首先,根据亲疏水氨基酸的旋转半径差来指导种群的变异;然后,构建距离分布模型和疏水概率模型来指导种群的更新,进而提高预测的精度.DHMA算法通过回转半径和疏水概率模型,有效的增强了疏水性在蛋白质的折叠过程中的驱动;同时通过构建距离分布模型,进一步加强了远程作用力的引导.10个测试蛋白的实验结果表明,DHMA算法具有较好的搜索性能和预测精度,是一种有效的构象空间搜索算法.
在下一步的研究中,我们将利用机器学习以及数据挖掘技术来预测高质量的距离谱,并且将其与疏水特性融合来改进现有的能量函数,以期得到更好的预测结果.同时,在以后的研究中,将会采用其他优化方法来进一步提高预测精度,如多目标优化技术和多模态优化方法.
