桑杞胶囊中总蒽醌含量测定的研究※
2020-01-10李钦青董虹玲
李钦青 董虹玲 张 海 张 力
(1 山西中医药大学中药学院,山西 晋中 030619;2 山西仁源堂药业有限公司,山西 太原 030024)
桑杞胶囊中主要含有决明子、枸杞子、桑椹、牛磺酸、葡萄糖酸锌、葡萄糖酸亚铁等药味组成,具有缓解视疲劳的作用。目前用于总蒽醌类成分含量测定的方法较多[1-8],但由于实验操作方法的差异,导致测定结果偏差较大,因此本实验对桑杞胶囊中总蒽醌的含量测定方法进行了研究,建立一个简便、准确的总蒽醌类成分含量的测定方法,为其质量控制提供参考。
1 仪器与材料
1.1 主要仪器TU-1810 紫外可见分光光度计(北京普析通用仪器有限责任公司)、METASH UV-6100S 紫外可见分光光度计(上海元析仪器有限公司)、FB224 自动内校电子分析天平(上海舜禹恒平科学仪器有限公司)、AB135-S 十万分之一电子天平(METTLER TOLEDO)。
1.2 药材与试剂 自制桑杞胶囊(批号20150701、20150702、20150703);1,8-二羟基蒽醌、标准品(中国药品生物制品检定所0829-9702);甲醇、乙醚、醋酸镁、盐酸、醋酸、过氧化氢均为分析纯。
2 方法与结果
2.1 显色剂的配制 醋酸镁甲醇溶液:称取适量醋酸镁,用甲醇溶解配制成0.5% 醋酸镁甲醇溶液,充分摇匀后备用。混合碱溶液:10% NaOH+4% 氨水等体积混合
2.2 对照品溶液的制备 称取1,8-二羟基蒽醌标准品13.00 mg,置50 mL 量瓶中,加入甲醇至刻度,摇匀,即得1,8-二羟基蒽醌对照品溶液(0.26 mg/mL)。
2.3 标准曲线的绘制 醋酸镁显色标准曲线的绘制:吸取1,8-二羟基蒽醌标准溶液0.05,0.10,0.20,0.40,0.60,0.80,1.00 mL(相当于1,8-二羟基蒽0.013,0.026,0.052,0.104,0.156,0.208,0.26 mg)分别置于10 mL 量瓶中,加0.5% 醋酸镁甲醇溶液至10.0 mL[9],摇匀,用分光光度计在523 nm 波长处以试剂空白溶液为参比,1 cm 比色皿测定吸光度值。以1,8-二羟基蒽醌浓度为横坐标,吸光度值为纵坐标,绘制标准曲线。得浓度-吸光度关系的回归方程:A=30.13C+0.172,r=0.99905,结果见表1。

表1 以0.5%醋酸镁甲醇为显色剂的标准曲线的制备
混合碱显色的标准曲线的绘制:精密吸取1,8-二羟基蒽醌标准溶液0.05,0.10,0.20,0.40,0.60,0.80,1.00 mL(相当于1,8-二羟基蒽0.013,0.026,0.052,0.104,0.156,0.208,0.26 mg)分别置于10 mL 量瓶中,蒸干甲醇,用混合碱溶液定容至刻度,置于暗处30 min,于520 nm 处测定吸光度。绘制标准曲线,得浓度-吸光度关系的回归方程:Y=29.761x+0.157 9,r=0.999,结果见表2。
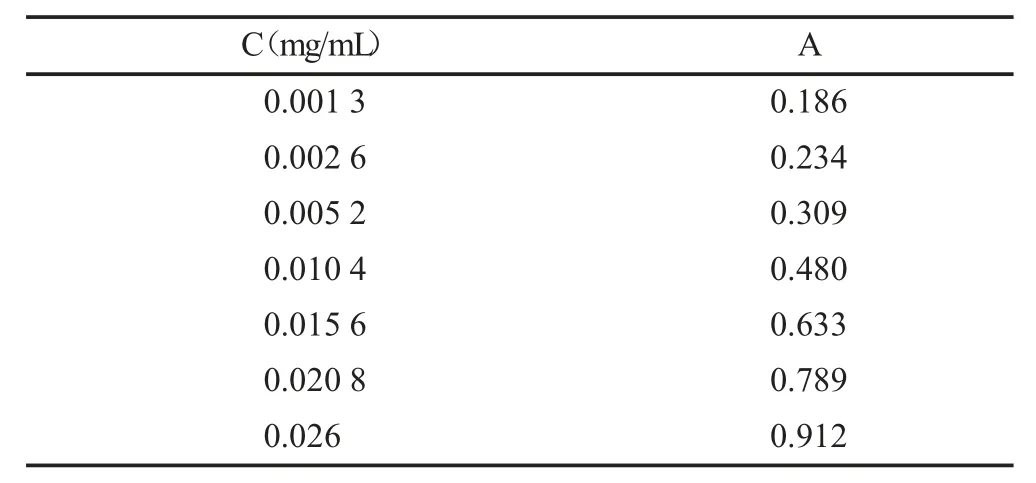
表2 混合碱显色的标准曲线的绘制制备
2.4 总蒽醌供试品溶液的制备 方法1:取胶囊内容物约2.0 g,精密称定。置锥形瓶中,准确加入甲醇50 mL,称定重量,90 ℃水浴回流1 h,放冷,称定重量,用甲醇补足减失的重量,滤过,精密移取续滤液10 mL,蒸干,加入40 mL 水溶解,再加入6 mL 过氧化氢(30%)、1 mL 盐酸(1+1),90 ℃水浴回流30 min,放冷,用乙醚提取三次,分别用量20 mL、20 mL、15 mL,合并乙醚液,用水洗2 次,每次用水量为10 mL,弃去水洗液。挥干乙醚,甲醇定容至25 mL,得待显色的样品溶液。显色方法(1):精密吸取5 mL 样品溶液,蒸干甲醇,用混合碱定容至10 mL 显色(置于暗处30 分钟)测定吸光度。显色方法(2):精密吸取5 mL 样品溶液,蒸干甲醇。用0.5% 醋酸镁甲醇溶液定容至10 mL,显色1 h 测定吸光度。把测定值代入回归方程,并计算其含量,实验结果见表3。
方法2:取胶囊内容物2.0 g,精密称定,置于250 mL 圆底烧瓶中,加混合酸(冰醋酸-25% HCl,18∶2)19.2 mL 在沸水浴中回流15 min,加乙醚提取3 次,每次10 mL,残渣再加入混合酸12.8 mL,在沸水浴中回流15 min,放冷,用乙醚提取3 次,每次10 mL 合并乙醚液于分液漏斗中,分别用水60 mL、50 mL 振摇两次,弃去水洗液,乙醚液蒸干,甲醇定容至25 mL,得待显色的样品溶液。显色方法同方法1,实验结果见表3。
方法3:取胶囊内容物2.0 g,精密称定,加少量水溶解(20 mL),加5 mol/L 硫酸20 mL,沸水浴回流2 h,放至室温,用二氯甲烷萃取5 次,每次10 mL,合并二氯甲烷,定容至25 mL,得待显色的样品溶液。显色方法同方法1,实验结果见表3。
方法4:取胶囊内容物2.0 g,精密称定,置于圆底烧瓶中,精密加入甲醇50 mL,称定质量,置水浴上加热回流30 min,放冷,再称定质量,用甲醇补足减失的量,摇匀,滤过。精密量取续滤液25 mL,蒸干,加10%(V/V)盐酸溶液30 mL,置沸水浴中加热水解1 h,立即冷却。用二氯甲烷强力振摇萃取4 次,每次20 mL,合并二氯甲烷液并回收至干。残渣用甲醇溶解,并定容至25 mL,摇匀,得待显色的样品溶液。显色方法同方法1,实验结果见表3。
方法5:取胶囊内容物2.0 g 精密称定,加水30 mL溶解,加入盐酸调至pH=1,沸水浴回流水解30 min,冷却,于分液漏斗中,用氯仿30 mL、20 mL、20 mL、20 mL 分次萃取,合并氯仿萃取液。用水30 mL、20 mL分次洗涤氯仿萃取液,弃去水液,氯仿液用蒸干,用甲醇溶解定容至25 mL,得样品溶液。显色方法同方法1,实验结果见表3。

表3 桑杞胶囊中总蒽醌含量测定结果
最终确定样品的处理方法为:称取混合均匀的胶囊内容物2.0 g,置于150 mL 锥形瓶中,加甲醇50 mL,于90 ℃水浴上加热1 h,放冷,滤过,精密移取试样提取液10.0 mL,置于150 mL 锥形瓶中,蒸干,残渣加水20 mL 使溶解,加3.0 mL 30% 过氧化氢溶液、0.5 mL 盐酸溶液(1+1),于90 ℃水浴上回流30 min,放冷,用乙醚提取3 次,每次20 mL,合并乙醚液,用水洗涤2 次,每次10 mL,乙醚液挥干,残渣用醋酸镁甲醇溶液溶解[10]并转移至10 mL 容量瓶中,加醋酸镁甲醇溶液稀释至刻度,混匀,作为供试品溶液。
2.5 精密度实验 取1,8-二羟基蒽醌对照品溶液(0.015 6 mg/mL)。0.5% 醋酸镁甲醇显色连续六次测定吸光度A,精密度良好,结果见表4。
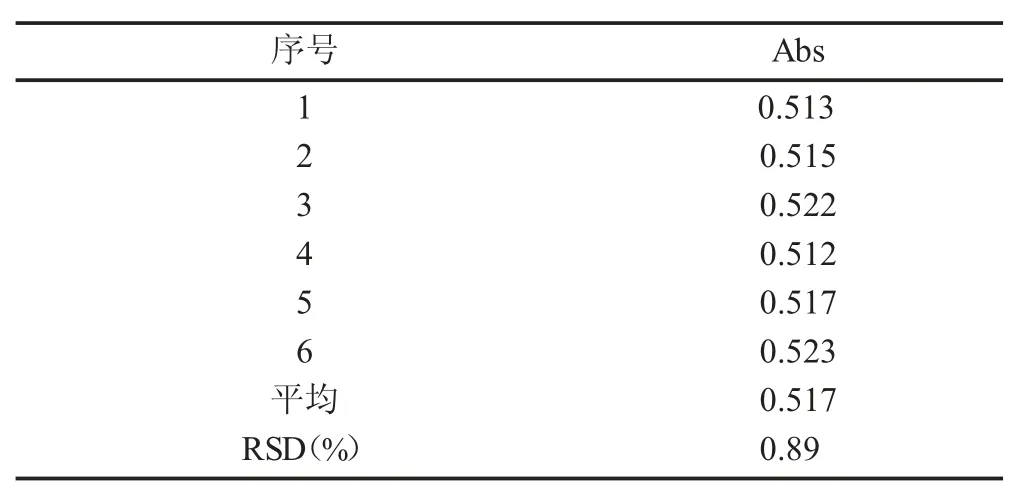
表4 精密度实验
2.6 稳定性实验 取胶囊内容物2.0 g,按供试品溶液制备方法制备供试品溶液,分别于10、30、60、90、120、180 min 测定吸光度,结果表明在3h 内稳定性良好,RSD 为2.85%,结果见表5。

表5 显色稳定性实验
2.7 重复性实验 取20150701 批样品内容物2.0 g,按照“供试品溶液的制备”项下的方法制得供试品溶液6 份,测定吸光度,计算含量,结果表明重复性较好,结果见表6。

表6 重复性实验
2.8 回收率考察 精密称取已知含量的样品1 g,按照供试品制备方法各供试液备用,取试样2 mL(测得试样中总蒽醌浓度为8.312 μg/mL)。分别加入1,8-二羟基蒽醌对照品溶液0.1 mL、0.2 mL、0.5 mL,用0.5% 醋酸镁甲醇溶液定容至5 mL 显色测定吸光度,结果见表7。
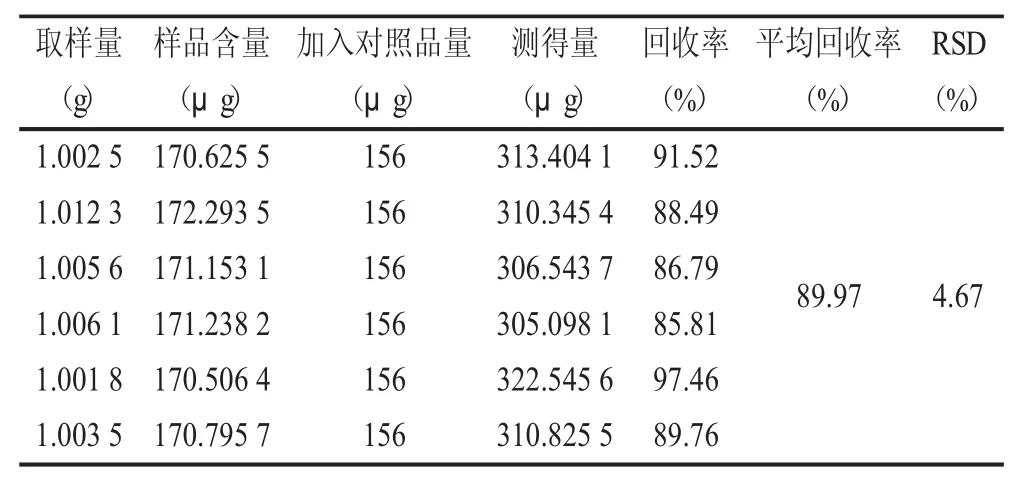
表7 回收率实验
2.9 含量测定 按照拟定实验方案测定三批自制桑杞胶囊中总蒽醌的含量,结果见表8。

表8 含量测定结果
3 讨论
3.1 提取溶剂的选择 方案中提取溶剂分别采用甲醇、70% 乙醇、乙醚、二氯甲烷、氯仿结果表明各种溶剂的提取率相似,从提取溶剂的挥发率、毒性大小以及提取效果的稳定性考虑甲醇为较优的提取溶剂,且文献中使用甲醇也较普遍。
3.2 酸水解的选择 方案中分别采用不加酸、混合酸溶液、5 mol/L 硫酸、盐酸这三种酸与不加酸的提取效果的对比,结果表明:混合酸与乙醚难分层,不适用,硫酸提取效果比盐酸好,但是硫酸显色与标准系列有差异,故不选用,所以选择盐酸作为酸水解的条件。
3.3 酸水解形式的选择 样品经过甲醇或其他溶剂提取之后再酸水解与直接进行酸水解的结果进行比较:直接酸水解提取的杂质较多,得到的样品溶液多呈红棕色,与标准品溶液的色差较大,对测定会产生一定的影响,样品在经过甲醇或其他有机溶剂处理后再经过酸水解可能会造成一部分结合蒽醌损失,所以测定结果偏小,但是本实验中测定的为胶囊内容物,在制作工艺中已经经过提取,损失成分微小,对结果影响不大,因此采用先甲醇提取再酸水解为较优条件。
3.4 氧化剂的选择 实验中加氧化剂与不加氧化剂的结果进行比较:加入氧化剂样品溶液显色较稳定,测定结果偏差小于未加入氧化剂的样品提取溶液。因为过氧化氢具有氧化和使部分杂质褪色的作用,使测定结果更准确、稳定。故选择加入氧化剂过氧化氢。
3.5 显色剂的选择 实验中对混合碱(10% NaOH+4% 氨水)与0.5% 醋酸镁甲醇的显色效果进行对比,醋酸镁甲醇比混合碱的显色效果稳定且溶液配制简单。
4 结论
本实验比较了不同提取溶剂,不同酸水解,不同水解方式和不同显色剂对桑杞胶囊中总蒽醌含量的影响,筛选出桑杞胶囊总蒽醌最佳提取方法,为桑杞胶囊中总蒽醌类成分提取的样品处理方法提供科学依据。并进行了方法学考察,建立桑杞胶囊中总蒽醌的含量测定方法,为其质量控制提供了参考。
