Co-Mo共掺杂Al2O3电子结构及透射率的第一性原理计算
2020-01-10方文玉王晓雯
方文玉, 王晓雯, 郑 勤, 高 深
(1. 湖北医药学院公共卫生与管理学院, 十堰 442000; 2. 湖北医药学院基础医学院 ,十堰 442000;3. 湖北医药学院后勤处, 十堰 442000; 4. 武汉大学电气工程学院, 武汉 430079)
1 引 言
Al2O3是自然界中十分常见的材料,被广泛用于工业和生活领域. Al2O3存在多种同质异形体,分别为-Al2O3、-Al2O3和-Al2O3,其中-Al2O3是低温下最稳定的相,具有低电导率和高硬度的特点,是很好的介电材料,在表面和薄膜领域有着广泛的应用. 有研究表明,通过材料掺杂能够使材料的结构、光电性质、热学性质等发生变化[1-3]. 例如,伏春平等人[4]发现Si掺杂Al2O3可以使半导体的导带和价带发生移动,掺杂体系能隙变小. 王兴军等人[5].通过溶胶-凝胶法制备Er3+-Yb3+共掺杂Al2O3粉末,实验结果发现共掺杂Al2O3粉末具有中心波长为1.533 μm的光致发光(PL)特性. 近年来,掺杂Al2O3光学器件被运用到很多元器件[6].通过Eu、Tb、Ce、Ab和Mn掺杂Al2O3薄膜可用于平板显示器、光学放大器的电子器件[7-11];掺杂Tb-Zn可用来制作长余辉发光材料[12-13], 他们能够通过掺杂离子杂质能级与其他能级间激发-退激而发光. 因此,研究掺杂型Al2O3的电子结构与光学性质,对材料的机理研究有着重要意义. 目前,关于Co、Mo掺杂Al2O3的研究也有一些报道,例如,刘欣等人[14]通过溶胶凝胶法和浸渍法制备Co/Fe/Al2O3/cordierite催化剂,发现Co掺杂可以增强催化剂抗SO2和H2O的能力;张玉涵等人[15]通过原位红外光谱法发现, 在Mo/γ-A12O3催化剂中加入助剂Co对Mo吸附位起到显著改性作用. 以上都是实验研究,关于理论计算方面的研究尚未见报道.因此,本文以Co、Mo单掺杂及共掺杂Al2O3为研究对象,通过第一性原理计算分析掺杂体系的电子结构及透射率,为新的功能材料研究提供理论指导.
2 模型结构和计算方法
2.1 结构模型
计算中选取α型Al2O3晶胞模型,其对称群为R-3C,晶格常数为a=b=4.759 Å,c=12.991 Å,α=β=90°,γ=120°. 掺杂时用Co、Mo替换晶胞中的Al原子,替换的位置如图1所示.
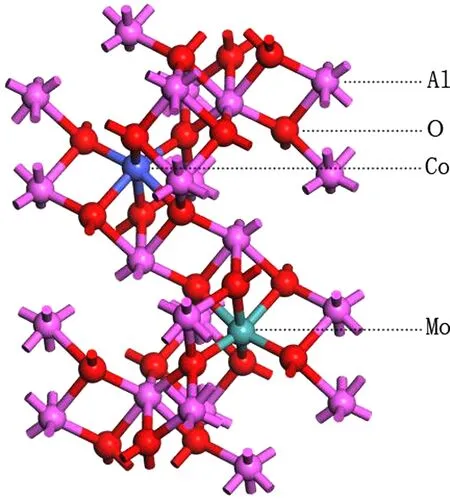
图1 Co-Mo共掺杂Al2O3晶胞模型
2.2 计算方法
本文计算是由CASTEP软件来完成,基于密度泛函理论[16]结合平面波赝势方法的计算程序,采用周期性边界条件,运用广义梯度近似GGA的PBE泛函计算方法来处理电子间的交换关联能.
选取价电子组态分别为Al-3s23p1、O-2s22p4、Co-3d74s2和Mo-4s24p64d55s1. 计算时选取1×1×1的Al2O3的超晶胞掺杂母体,平面波截断能设置为Ecut=450 eV,布里渊区积分采用3×5×2 Monkhorst-Pack特殊K点对全布里渊区求和,计算均在倒格矢中进行. 在自洽场计算中,能量的收敛精度为1.0×10-5eV/atom,每个原子上的受力不大于0.05 eV/nm,内应力收敛精度0.05 GPa,原子最大位移收敛标准1.0×10-3nm,计算各种体系时均进行结构优化.
3 计算结果与讨论
3.1 晶体结构
表1是本征及掺杂Al2O3优化后的晶格常数及掺杂结合能,可以发现掺杂后晶胞的晶格常数及体积均在增加. 这是因为Co(0.072 nm)和Mo(0.062 nm)的离子半径大于Al(0.053 nm)离子半径,形成的Co-O键和Mo-O键比Al键更长,同时,Co和Mo替换Al后,使得离子多余正电荷之间相互排斥力增大,系统能量增高而导致晶胞体积进一步增大[17].
为了表征离子掺杂难易程度,引入掺杂结合能的概念,在电子在非自旋极化条件下,掺杂结合能的表达式[18]:
Ef=ET-EAl2O3-Ex+EAl
(1)
其中,Ef表示掺杂体系结合能,ET表示掺杂后体系的总能量,EAl2O3表示本征Al2O3总能量,Ex和EAl分别表示单个掺杂原子和Al原子的能量. 从表1可以看出,Co单掺杂的结合能最大,最不容易掺杂;而Mo掺杂的结合能最低,最容易掺杂,掺杂后的Al2O3最稳定.
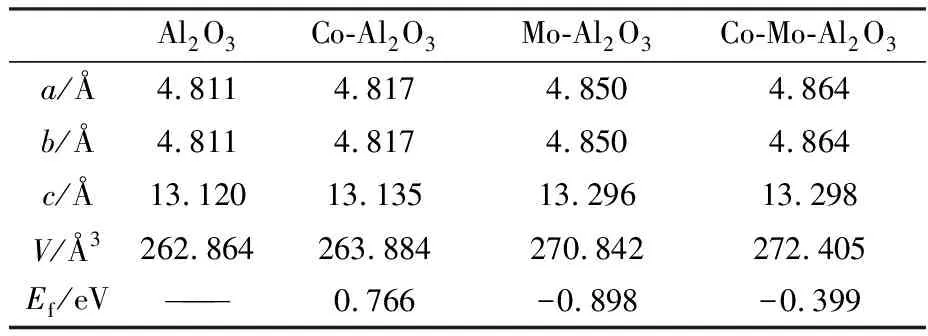
表1 优化后的晶格常数和结合能
3.2 能带结构和态密度分析
图2是本征及掺杂Al2O3能带图,为便于分析,选取费米能级附近(-6~12 eV)的能带结构,图3是本征及掺杂Al2O3的分态密度,这里选取-20~10 eV区间. 从图2(a)可以发现,本征Al2O3的价带顶部和导带底部都出现G点,说明Al2O3是直接带隙半导体,其禁带宽度Eg=5.96 eV,这与高丽红等人[19]的计算结果基本一致. 通过观察图3(a)发现,导带主要由Al-3s和Al-3p组成,价带则分为两个区域,上价带(-7~0 eV)主要由O-2p态电子组成,下价带(-18~16 eV)则由O-2s态电子组成. 从图2(b)、(c)可知,与本征Al2O3相比,Co、Mo单掺杂Al2O3导带结构与之相似,价带区域平缓,价带和导带均发生下移,并且在禁带中引入杂质能级,费米能级相对靠近导带,远离价带,属于n型掺杂. Co-Mo共掺杂后,杂质能级变得更加丰富,同时具有Co、Mo单掺杂的能带特征. 通过观察掺杂后的分态密度发现,杂质能级主要由Co-3d态电子和Mo-4d态电子组成,且掺杂原子态密度在费米面附近均存在一个峰值,这能够增加掺杂体系的载流子浓度,改善Al2O3的导电性,因此Co、Mo是制备低阻Al2O3的合适掺杂元素. 此外,Mo单掺杂Al2O3时,Mo-4d态电子在费米面以下主要由两部分组成,分别分布在-11~-5 eV和-1~0 eV,这两部分电子与O-2p态电子相互作用,使得O-2p态电子也劈裂为两部分,由此产生禁带. 同样,共掺杂时,Mo-4d与Co-3d相互作用,Co-3d也会被分为两部分,同时,在费米面附近Co-3d态电子的增加,将会影响电子在各量子态之间的跃迁,这也是掺杂改变Al2O3光学性质的根本原因.
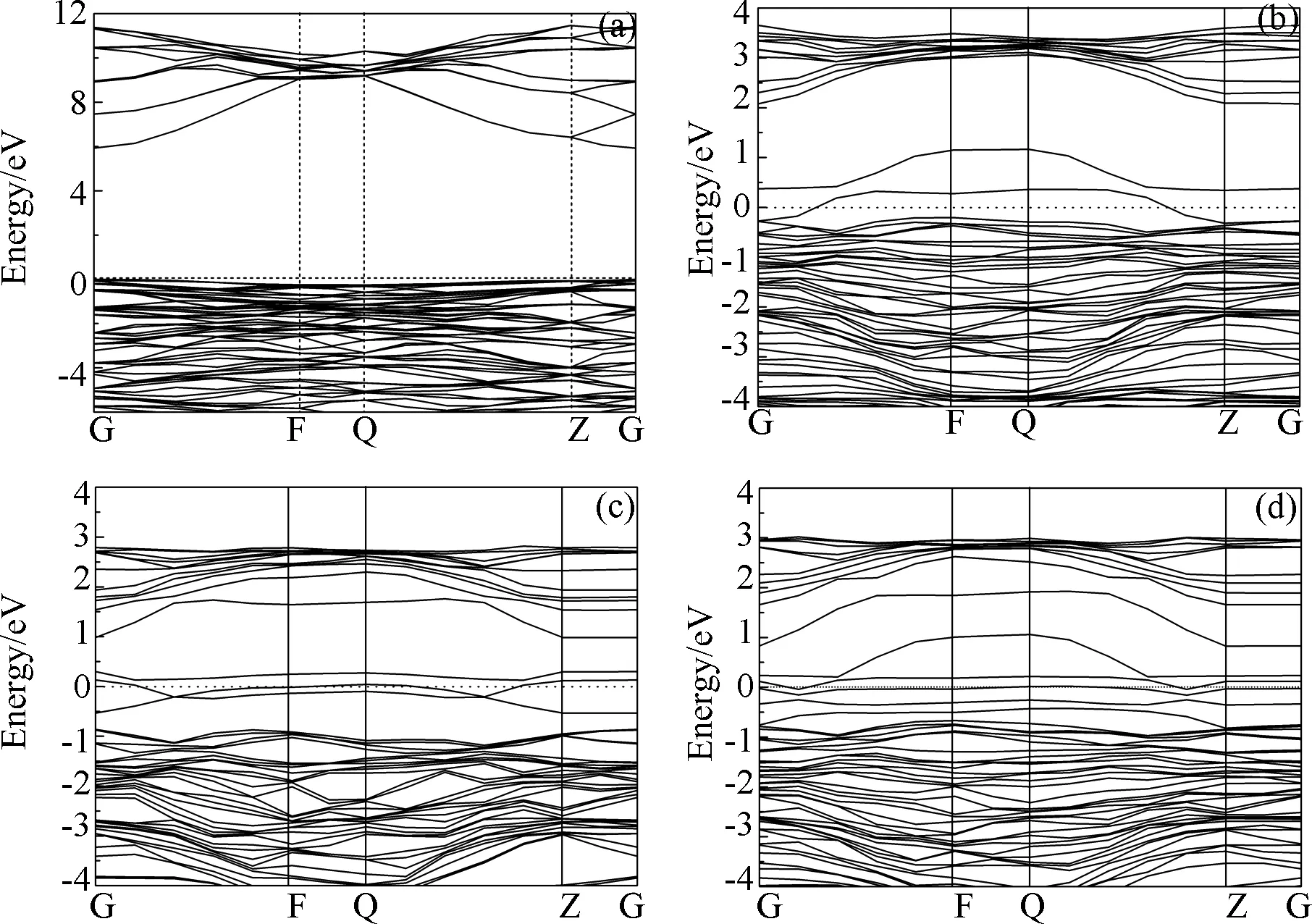
图2 本征及掺杂Al2O3能带图:(a)本征Al2O3;(b) Co掺杂Al2O3;(c) Mo掺杂Al2O3;(d) Co-Mo共掺杂Al2O3Fig.2 The band structures of (a) pure Al2O3; (b) Co doped Al2O3; (c) Mo doped Al2O3; (d) Co-Mo codoped Al2O3

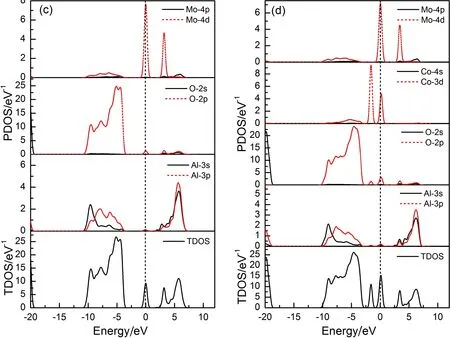
图3 本征及掺杂Al2O3分态密度: (a)本征Al2O3;(b) Co掺杂Al2O3;(c) Mo掺杂Al2O3;(d) Co-Mo共掺杂Al2O3
Fig.3 The DOSs of (a) pure Al2O3; (b) Co doped Al2O3; (c) Mo doped Al2O3; (d) Co-Mo codoped Al2O3
3.3 透射率
为研究Co、Mo单掺杂及共掺杂Al2O3的光学透射率,首先要计算各掺杂体系的吸收系数与反射率,计算时选取入射光的极化方向为<100>的垂直照射.
吸收系数α是指光子通过单位厚度的薄膜材料时能量的衰减分数. 半导体材料能级间的跃迁会造成光子能量的衰减,例如能带间的跃迁、带内的能级跃迁以及缺陷、掺杂、晶格振动等有关的跃迁过程都会对光子的吸收性能产生影响. 图4是本征及掺杂Al2O3的吸收系数与反射率,从图4(a)可以看出,相对于本征Al2O3,Co单掺杂的反射率没有太大变化,Mo单掺杂及Co-Mo共掺杂Al2O3的反射率在低能量区域(0~2 eV)有明显起伏,对可见光及远红外光谱有较明显的反射效果. 观察图4(b)发现,本征Al2O3的吸收边约为6 eV,这与它的禁带宽度5.96 eV非常接近,这是因为吸收边与光学带隙直接相关.由于光-电子的耦合效应会引起电子在价带与导带间跃迁,其中最小的跃迁能量起点应为禁带宽度,因此Al2O3半导体的吸收边应该大于或等于带隙,即6>5.96 eV. 掺杂后,体系的吸收边发生红移,通过图2和图3可知,这主要是因为掺杂原子的d态电子形成的杂质能级拉近了能级之间的距离,电子在能级之间跃迁变得更容易,吸收光子的能量也小于本征Al2O3的禁带宽度. 同时,通过观察吸收系数的峰值可以发现,掺杂体系分别在1、 4和8 eV附近出现峰值,通过观察分态密度可知,第一个峰值来自Mo-4d态电子价带与导带之间跃迁,第二个峰值则是由价带中O-2p态电子向费米面上的Co-3d态电子跃迁产生,最后一个峰值则是由价带O-2p态电子向费米面上Mo-4d态电子跃迁产生的.
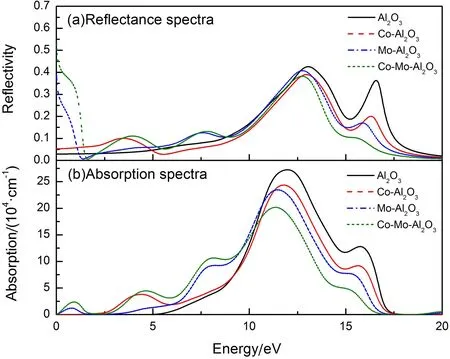
图4 本征及掺杂Al2O3光学性质: (a)反射率;(b)吸收系数
Fig. 4 The optical properties of intrinsic and doped Al2O3: (a) reflectivity; (b) absorption
假设光子照射到厚度为d的薄膜,忽略其光干涉效应,则透射率T可表达为[20]:
(2)
文献[6]通过中频反应磁控溅射制备Al2O3薄膜,选取的厚度平均值为650 nm.为了便于比较,计算时设薄膜厚度d=650 nm,计算得到本征及掺杂情况下Al2O3薄膜的光学透射率如图5所示.
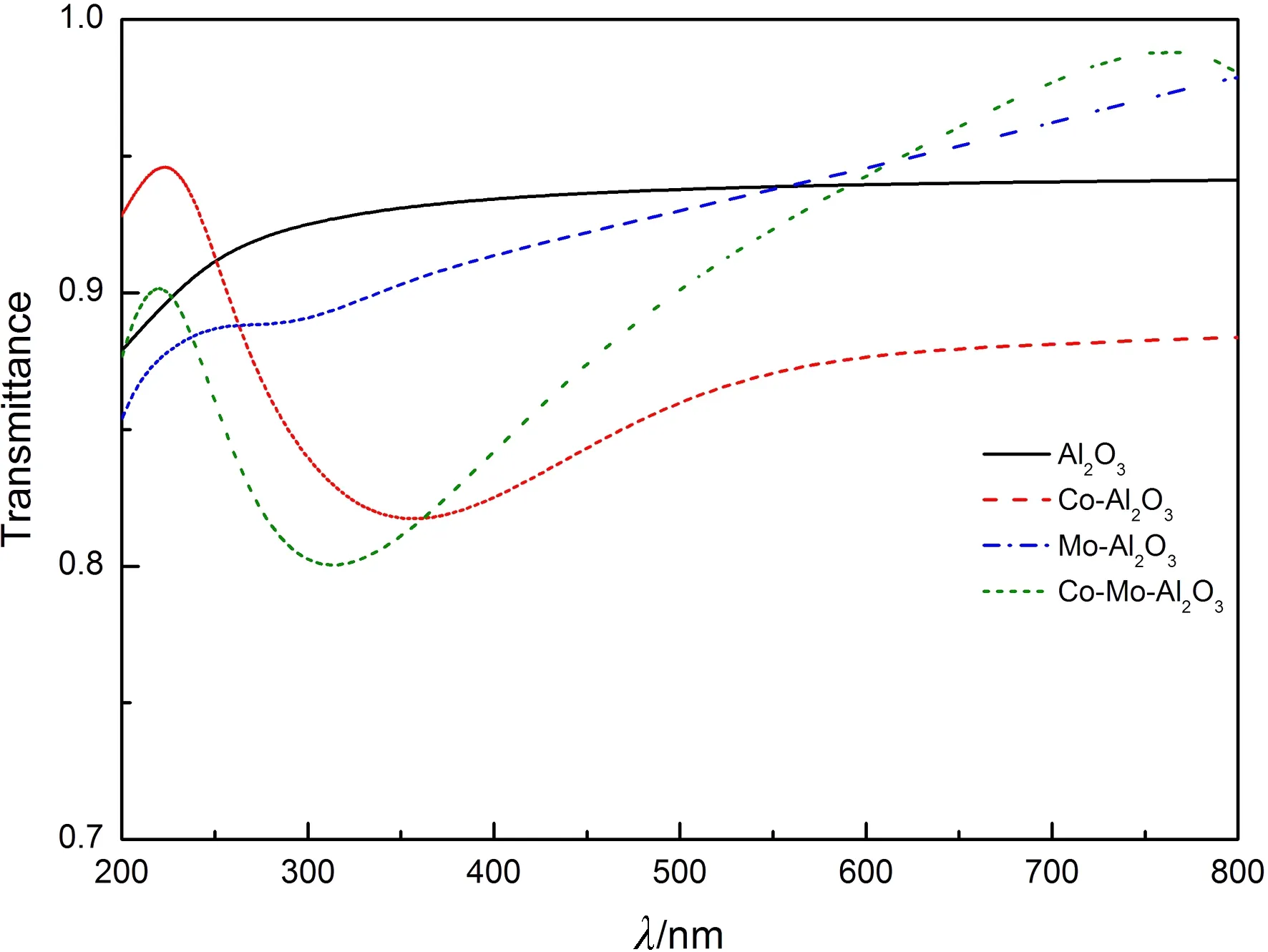
图5 本征及掺杂Al2O3的透射率
Fig.5 The transmittances of intrinsic and doped Al2O3
从图5可以看出,本征Al2O3薄膜在200~800 nm范围内的透射率约为92%,与实验结果相符. 根据能量守恒定律,光子照射物体时,分为吸收光、反射光、透射光三部分. 掺杂后,Al2O3吸收边发生红移,因此在可见光和近红外(380~780 nm)的平均透射率减小,其中Co单掺杂的透射率下降的最明显. 但是在600~780 nm波段内,Mo单掺杂和Co-Mo共掺杂的透射率逐渐增大,在93%~98%之间. 另外,在200~280 nm的段波紫外光区域,Co单掺杂的透射率相对较大,最高可达95%.
4 结 论
采用基于密度泛函理论(DFT)的第一性原理平面波超软赝势方法,计算了本征Al2O3,Co、Mo单掺杂以及Co-Mo共掺杂Al2O3的电子结构和光学性质. 计算结果表明:Mo单掺杂以及Co-Mo共掺杂Al2O3的结合能较低,比较容易合成. Co、Mo掺杂均属于n型掺杂,能够提升掺杂体系的载流子浓度,改善Al2O3的导电性. 此外,掺杂体系的杂质能级主要由Co-3d态电子和Mo-4d态电子组成,这些杂质能级拉近了级之间的距离,电子在能级之间跃迁变得更容易,这是影响掺杂Al2O3光学性质的内在原因. 掺杂后,吸收光谱发生红移现象,且光学性质变化主要集中要低能量范围. 本征Al2O3薄膜在200~800 nm范围内的透射率约为92%,Mo单掺杂和Co-Mo共掺杂Al2O3在600~780 nm波段内的透射率达到93%~98%,在200~280 nm的短波紫外光区域,Co单掺杂的透射率相对较大,最高可达95%. 因此,三种掺杂Al2O3适用于制备各类光学透射膜.
