驱动蛋白超家族作为肺癌潜在生物标志物和治疗靶点的研究进展
2020-01-07马宇星梁艺颖王誉熹
杨 阳,马宇星,梁艺颖,王誉熹
(1.四川大学华西口腔医学院,成都 610041; 2.四川大学华西公共卫生学院,成都 610041;3.四川大学华西医院呼吸与危重症医学科,成都 610041)
肺癌是全球发病率和病死率最高的恶性肿瘤之一,占癌症总发病人数的11.6%、总死亡人数的18.4%[1]。肺癌可分为非小细胞肺癌和小细胞肺癌两大类,非小细胞肺癌可进一步细分为几种组织学亚型,其中鳞状细胞癌和腺癌最多见。晚期非小细胞肺癌常采用含铂化疗和局部放疗相结合的方法治疗,但其患者5年生存率仅为5%~15%,且目前没有得到显著改善;而小细胞肺癌是侵袭性的神经内分泌恶性肿瘤,易迅速复发,患者5年生存率不到5%[2]。被批准用于小细胞肺癌的药物只有依托泊苷和拓扑替康,虽然初始反应有60%~80%,但由于癌症干细胞样亚群导致多重耐药性[3],药效往往非常短暂且极易复发。目前临床上肺癌诊断的金标准是病理穿刺检查,但穿刺活组织检查为有创性操作,患者接受程度较低;影像学检查及糖类抗原19-9、血清癌胚抗原等肺癌相关抗原检测的敏感性与特异性相对较弱,只能作为辅助诊断或筛查指标。而驱动蛋白在肺癌的发生发展中起重要作用,不仅在肿瘤细胞染色体的运动及异常分裂、侵袭和转移中发挥重要作用,还能影响胞内增殖凋亡信号通路,是非常有潜力的肺癌生物治疗靶点。此外,驱动蛋白还能作为肺癌的诊断和预后标志物,为治疗方案的拟定提供肿瘤分期和预后信息。现就驱动蛋白超家族(kinesin superfamily proteins,KIFs)作为肺癌潜在生物标志物和治疗靶点的研究进展进行综述。
1 驱动蛋白概述
驱动蛋白于1985年首次被发现,是从乌贼神经轴突和视叶中分离出的一类分子马达,其能够利用腺苷三磷酸酶水解所释放的能量驱动自身及所携带的物质分子沿微管细丝正极做定向运动,进行胞内物质(如信息分子、囊泡、细胞器、染色体、蛋白质复合体)和信使RNA的运输,并调节信号通路,从而发挥生物学效应[4],对细胞的功能和形态至关重要。
目前KIFs有45个成员,被分为14个亚家族,命名为kinesin1~kinesin14,其中38种表达于神经组织[5]。驱动蛋白分子由两条重链和两条轻链组成,包括马达结构域、PH结构域和FHA结构域,其中马达结构域具有腺苷三磷酸结合位点及微管结合位点,并在KIFs中高度一致[6]。根据马达结构域在重链上的位置不同,可分为在C端的C-kinesin、N端的N-kinesin以及中间的M-kinesin,大多数的驱动蛋白属于N-kinesin。通常,N-kinesin和C-kinesin分别驱动微管正末端和负末端定向运动,而M-kinesin负责解聚微管[7]。PH结构域参与货物的运输,且能够决定结合货物的特异性,但FHA结构域的功能仍然未知。驱动蛋白不能单独发挥作用,了解驱动蛋白的关键是研究其与货物的连接机制,通过将货物与驱动蛋白进行特定配对,可以对其相互作用进行微调[8]。
近年来,驱动蛋白的作用已从内在细胞过程扩展到病毒的发病机制,包括单纯疱疹病毒和痘苗病毒[9]。此外,驱动蛋白活性与神经元健康及功能联系密切,特别是神经元依赖于驱动蛋白进行长距离转运以及突触小泡前体和神经递质受体(如谷氨酸)的正确定位[10]。驱动蛋白与一些神经发育或退行性疾病(如肌萎缩侧索硬化和遗传性痉挛性截瘫)相关。然而目前有许多KIFs成员的功能尚未阐释清楚,未来深入研究驱动蛋白与货物的结合运输机制以及信号系统的调节作用,可能能更好地理解驱动蛋白在疾病发生、发展中的功能。
2 驱动蛋白在肺癌中的作用
近年来研究表明,驱动蛋白表达水平的变化与肺癌的发生发展有直接关联,驱动蛋白异常可以通过染色体过度凝集、纺锤体形成异常、细胞分裂缺陷、形成后期桥或非整倍体及有丝分裂阻滞,改变细胞内遗传物质的分布,使细胞周期失控,最终渐进性地导致肿瘤发生[11]。不同驱动蛋白亚家族成员作用机制稍有不同(见表1)。
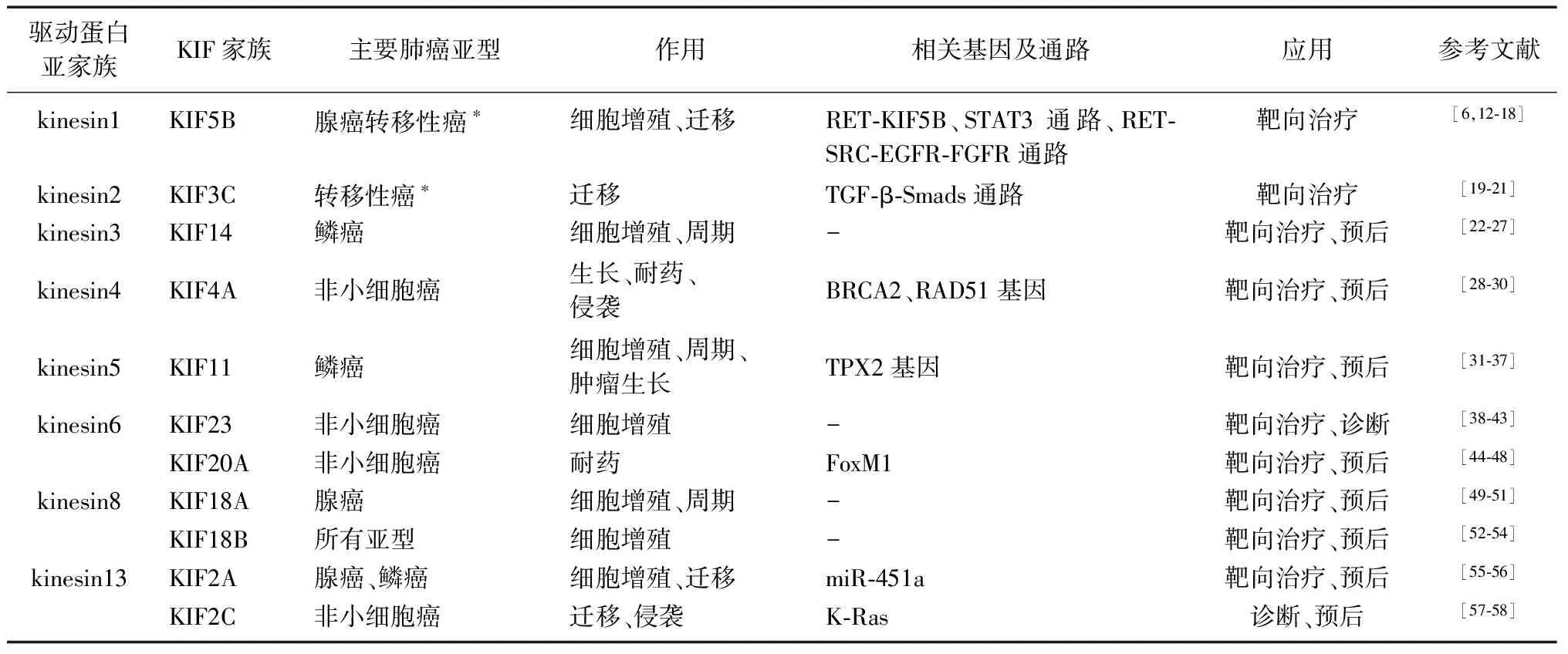
表1 驱动蛋白亚家族在肺癌中的作用
KIF:驱动蛋白家族;RET:转染重排;STAT3:信号转导及转录激活因子3;SRC:类固醇受体共激活因子;EGFR:上皮生长因子受体;FGFR:成纤维细胞生长因子受体;TGF-β:转化生长因子-β;BRCA2:乳腺癌易感基因2;RAD51:DNA修复相关蛋白51基因;TPX2:Xklp2靶蛋白基因;FoxM1:叉头盒蛋白M1基因;K-Ras:一种原癌基因;*乳腺癌转移性肺癌,未提及病理类型;-:未报道
2.1kinesin1 KIF5B基因位于第10号染色体 p11.22区,表达产物KIF5B蛋白为kinesin1的一员,由9 634个氨基酸组成,在胞内运输、有丝分裂、细胞形成等方面起重要作用[6]。转染重排(rearranged during transfection,RET)基因重排是肺癌发生的驱动因素,最常见的重排为KIF5B-RET(72%),被认为是导致肺癌的主要基因融合类型,大多数KIF5B-RET重排患者从不吸烟(63%),多为肺腺癌(98%)及晚期患者(91%)[12]。Dacic等[13]认为,RET-KIF5B融合可能是放射性肺腺癌的一种遗传机制。Qian等[14]认为,KIF5B-RET融合激酶通过多水平活化信号转导及转录激活因子3促进肺癌细胞生长。而Das和Cagan[15]证明,KIF5B-RET的驱动蛋白和激酶结构域共同作用建立了微管和囊泡依赖性的RET-类固酶受体共激活因子-上皮生长因子受体-成纤维细胞生长因子受体信号转导中枢。KIF5B除对肺癌的发生起重要驱动功能外,对肿瘤肺转移也有重要转运作用。Wang等[16]发现,磷脂酶D2产生的磷脂酸能与KIF5B结合,促进膜型基质金属蛋白酶1表面转运,从而促进小鼠乳腺癌细胞的肺转移。Drilon等[17]进行的Ⅱ期临床试验证明,RET抑制剂卡博替尼能在RET重排的肺癌患者中产生快速和持久反应,但需要减少剂量。这可能与抑制血管内皮细胞生长因子受体2和其他非RET激酶而引起的目标外毒性有关[18]。单独抑制RET的药物在KIF5B-RET转化细胞中表现不佳,但将RET抑制剂索拉非尼与靶向上皮生长因子受体、微管或成纤维细胞生长因子受体的药物相结合,在果蝇和人细胞系KIF5B-RET模型中均具有强效[15]。
2.2kinesin2 KIF3C能够与KIF3A蛋白结合,是kinesin2的重要组成部分[19]。据报道,KIF3C是一种损伤特异性的激酶,它通过调节和组织生长锥中的微管细胞骨架来促进轴突的生长和再生[20]。驱动蛋白是Smad2磷酸化和转运所必需的物质,而转化生长因子-β(transforming growth factor-β,TGF-β)/Smads信号在乳腺肿瘤转移过程中起重要作用。Wang等[21]进行体外实验,发现TGF-β1诱导的磷酸化Smad2水平在乳腺癌细胞系中被KIF3C抑制剂作用后显著降低,乳腺癌的肺淋巴结转移被抑制,证明KIF3C在Smad2磷酸化和(或)转运中起作用,且可以通过抑制KIF3C来控制Smad2磷酸化,从而抑制乳腺癌细胞中的TGF-β信号转导,进而抑制乳腺癌的肺转移。
2.3kineisin3 KIF14于1994年被发现,是kinesin3的家族成员[22]。KIF14定位于HeLa细胞的中心纺锤体,对于胞质分裂的最后阶段至关重要,KIF14与香橼激酶的蛋白质调节剂相互作用,在胞质分裂中的中间体有中心组织作用;敲除KIF14会导致中期平板染色体错位,延迟有丝分裂、双核细胞出现,从而导致多倍体和细胞凋亡[23]。癌症的进展始于基因组的改变,其次反映在基因表达的改变上,有研究发现,KIF14是包括肺癌在内的多种癌症的候选癌基因[24]。在肺癌中发现了含有KIF14的1q32的最小增益区域,该区域的增益与癌前病变至恶性病变的进展以及鳞状细胞肺癌的转移相关[25]。已有研究发现22例原发性肺肿瘤中有10例KIF14信使RNA表达增加3~34倍,在这个非常小的初步队列中,KIF14水平升高的患者显示出生存时间缩短的趋势[24]。Corson等[26]首次通过大规模研究得出,KIF14信使RNA的表达是肺癌无病生存预后的独立指标。高水平的KIF14可能通过刺激过早的胞质分裂和(或)逃避纺锤体检查点而促进不受控制的增殖和癌细胞的非整倍体[27]。
2.4kinesin4 KIF4A大多在间期与核基质相关,而其中一小部分位于细胞质,能够调节染色体的凝聚和分离,在后期纺锤体运动和胞质分裂中起重要作用[28]。通过基因芯片技术发现,KIF4A基因在肺癌临床标本和细胞系中过表达;临床病理证据表明,KIF4A阳性的非小细胞肺癌患者的肿瘤特异性生存期较KIF4A阴性患者短;多因素分析证实,KIF4A基因在手术切除标本中的过表达可作为预后不良患者辅助治疗的参考指标[29]。非小细胞肺癌患者通常使用以铂为基础的化疗药物(如顺铂),该药物通过DNA双链断裂等机制发挥抗癌作用,KIF4A对DNA双链断裂修复能力的增强有重要作用,可能与先天或获得性耐药有关;试验发现通过干扰小RNA去除KIF4A后,顺铂诱导的人非小细胞肺癌细胞周期阻滞于S期和G2/M期,细胞毒作用显著增强;而KIF4A抑制剂能够抑制顺铂诱导的人非小细胞肺癌患者的乳腺癌易感基因2和DNA修复相关蛋白51基因的病灶形成能力,并限制顺铂诱导的多聚ADP核糖聚合酶1活性的进一步增强;研究证实,KIF4A中的色激肽是一种新的顺铂敏感性调节剂,它通过多种机制在人非小细胞肺癌细胞中显著增强这种敏感性[30]。这些研究结果表明,KIF4A分子能够为抗癌药物的开发以及临床预后生物标志物的确定提供依据。
2.5kinesin5 KIF11是kinesin5家族的一员,在纺锤体中交叉连接反平行的微管[31]。KIF11能够控制微管的正确排列,防止微管的降解,被人类Xklp2靶蛋白基因编码的蛋白调控,在肺癌的发生发展中起重要作用[32]。在96%的非小细胞肺癌患者肿瘤组织中KIF11高表达,其中鳞状细胞癌中基因的过表达显著高于腺癌,与正常组织中的基因表达比较差异有统计学意义;免疫荧光研究表明,这种有丝分裂相关蛋白在细胞分裂过程中与纺锤体共定位,在单变量分析中,KIF11主要在肺腺癌中成为预后指标[33]。Kato等[34]发现,肺大细胞癌和鳞状细胞癌中KIF11高水平表达的患者总体生存时间远低于KIF11低水平表达者,认为KIF11的高表达是肺大细胞癌和鳞状细胞癌患者的独立预后因素。干扰小RNA敲除KIF11[34],或使用异松果苷、SB-743921、ARRY-520和氯丙嗪抑制KIF11,对肿瘤生长有显著抑制作用,均可导致非小细胞肺癌细胞株存活率显著下降[35]。Lee等[36]发现,喷他脒和氯丙嗪在有丝分裂中的双重作用可产生协同抗肿瘤作用。体外研究发现,白藜芦醇能够有效抑制A549细胞生长,通过分析基因和蛋白表达的变化发现KIF11下调,表明白藜芦醇可能对细胞分裂G2/M期也有调节作用[37]。
2.6kinesin6
2.6.1KIF23 KIF23是胞质分裂的关键调节因子,KIF23功能障碍导致胞质分裂不完全,形成双核或多核细胞,预示着肿瘤细胞的形成;而KIF23过表达可能导致染色体分离缺陷,从而导致染色体不稳定,形成非整倍体,与肿瘤发生密切相关[38]。KIF23在乳腺癌[39]、神经胶质细胞瘤[40]等多种肿瘤组织中表达上调,猜测KIF23可能在非小细胞肺癌的发展中起一定作用。Välk等[41]发现,KIF23在非小细胞肺癌中表达上调,Kato等[42]的研究得到相同的结论,且通过多变量分析,肺腺癌KIF23高表达也被确定为一个独立的预后因素。RNA干扰介导的KIF23的缺失抑制了肺癌细胞株体内的肿瘤形成和诱导细胞凋亡,表明KIF23可能是治疗肺癌的一种新靶点[43]。以上研究一致表明KIF23在非小细胞肺癌的发生发展中起重要作用,且可作为生物标志物和药物靶点指导非小细胞肺癌的诊断与治疗,但关于KIF23在肿瘤发生中的具体机制目前尚无有关研究进行阐释。
2.6.2KIF20A KIF20A又称有丝分裂蛋白样蛋白2,与高尔基体运动有关,在间期与结合鸟苷三磷酸的Rab6蛋白相互作用,是细胞分裂成功过程中细胞周期调控的关键[44]。KIF20A过表达与肿瘤细胞增殖、肿瘤进展及侵袭、临床疗效差、总生存率低相关,被认为是一种肿瘤相关抗原[45]。KIF20A在胰腺癌、胃癌、卵巢透明细胞癌等多种癌症的发生发展过程中发挥重要作用,在肺癌中的作用目前研究较少。Zhao等[46]的研究首次分析了KIF20A在肺腺癌中的表达及功能,他们通过免疫组织化学分析发现,与正常肺组织相比,肺腺癌组织中的KIF20A水平普遍上调;此外,KIF20A的高表达与更短的总生存期显著相关,多变量回归分析显示,KIF20A是肺腺癌的独立预后因素,沉默KIF20A后,细胞增殖受到抑制,且细胞凋亡增加。因此,KIF20A可能是肺腺癌新的潜在标志物。放疗可诱导多种肿瘤细胞转移,放射敏感性降低是癌症放疗过程中遇到的主要障碍之一。以往研究表明,叉头盒蛋白M1(forkhead box protein M1,FoxM1)可能是诱导肿瘤侵袭转移的关键基因,FoxM1高表达与非小细胞肺癌患者预后不良显著相关[47]。Xiu等[48]的数据表明,抑制FoxM1可提高肺癌细胞对放疗的敏感性;与单独放疗相比,剔除FoxM1基因后迁移和侵袭显著减少;研究还发现,KIF20A在肺癌放疗后表达增加,且KIF20A过表达显著增加了肺癌细胞的增殖、侵袭和迁移,提示KIF20A可能与放疗抵抗有关;由于KIF20A是FoxM1的转录靶点之一,他们进一步研究了KIF20A与FoxM1的关系,结果表明KIF20A过表达解除了FoxM1对细胞增殖的抑制作用,从而减少了非小细胞肺癌细胞A549的凋亡,提示KIF20A在放疗后FoxM1介导的肺癌细胞辐射抵抗中起重要作用。但如何通过KIF20A解决最终的放疗抵抗问题仍需进一步研究。
2.7kinesin8
2.7.1KIF18A KIF18A主要位于细胞质和细胞核内,由898个氨基酸残基组成,分子量为100 000,已被证明是微管正端定向解聚剂,在膜细胞器以及蛋白复合物的定位中扮演重要角色[49-50]。KIF18A与乳腺癌、肝癌等多种肿瘤的发生、发展和预后关系紧密。KIF18A在肺腺癌组织中的表达显著高于正常组织,KIF18A高表达的患者总体生存期和无复发生存期明显较差,KIF18A表达增加可作为乳腺癌的独立预后因素;通过对肿瘤基因组图谱肺腺癌深度测序数据的分析,在2.6%的肺腺癌病例中检测到KIF18A突变,KIF18A基因的下调在体内外均明显抑制细胞增殖,沉默KIF18A可诱导肺腺癌细胞凋亡[51]。上述研究表明,KIF18A能够促进细胞增殖,抑制细胞凋亡,是肺腺癌患者有价值的预后预测因子和潜在的治疗靶点。
2.7.2KIF18B 与KIF18A相同,KIF18B也限制了微管在有丝分裂过程中的生长。不同的是,KIF18B定位于正端依赖于与正端跟踪蛋白EB1的结合,这使得KIF18B潜在的正端定向运动与正端积累之间的关系尚不清楚。KIF18B基因敲除可导致纺锤体微管组织缺陷和星形小管数量急剧增加,KIF18B敲除细胞在染色体排列上有缺陷,但在染色体分离上无缺陷[52]。目前关于KIF18B在肿瘤中的作用研究甚少,但有证据表明,KIF18B促进了宫颈癌等肿瘤的发展进程[53]。Itzel等[54]的研究表明,KIF18B基因在肺癌组织中的平均表达是正常组织的22倍,KIF18B过表达可促进细胞增殖,并且基于数据库发现KIF18B与肺癌的预后有关。有关kinesin8家族蛋白的研究目前仍在数量和质量上较为缺乏,同其他蛋白一样,在影响肿瘤的分子机制方面几乎处于空白状态。
2.8kinesin13
2.8.1KIF2A KIF2A在细胞分裂过程纺锤体的形成及细胞运动过程骨架的改变中具有重要作用,其因在乳腺癌、胃癌、弥漫性大B细胞淋巴瘤等多种人类癌症中的致癌作用及预后价值而受到关注。Xie等[55]发现,与邻近正常组织相比,KIF2A在肺腺癌组织中过表达。 Uchida等[56]在肺鳞癌临床标本中证实了miR-451a的下调,并发现miR-451a的低表达与肺鳞癌患者的不良预后显著相关,而miR-451a能够直接调控KIF2A表达,多因素分析表明,KIF2A在肺腺癌组织中的高表达是影响肺腺癌患者生存的独立危险因素;在体外,KIF2A基因敲除可明显抑制上皮-间充质转化和肺腺癌细胞的迁移,沉默KIF2A可抑制肺腺癌细胞增殖和诱导细胞凋亡。因此,KIF2A可作为肺腺癌的一个有价值的预后指标和有前景的治疗靶点。此外,功能分析表明,KIF2A是肺鳞癌发病机制中的重要基因,其过表达参与肺鳞癌的发病过程,故KIF2A基因被定性为一种原癌基因[56]。
2.8.2KIF2C KIF2C可调节细胞中的微管运动,对有丝分裂后期染色体分离非常重要。Huang和Gao[57]发现,KIF2C在非小细胞肺癌与邻近正常组织中存在差异表达,KIF2C信使RNA水平在非小细胞肺癌组织和细胞中上调;且多变量分析表明,KIF2C在肺腺癌中的过度表达是肺腺癌患者总体生存率较低的独立危险因素;进行生存分析评估发现,KIF2C与非小细胞肺癌的预后密切相关,KIF2C可能成为非小细胞肺癌患者潜在的诊断和预后生物标志物。关于KIF2C在肺癌细胞中的调节机制,Zaganjor等[58]认为,由于KIF2C的解聚活性增加了微管的动态不稳定性,从而影响K-Ras(一种原癌基因)突变,上调细胞迁移和侵袭基质凝胶的能力,以增强肿瘤细胞的迁移和侵袭。
3 小 结
近年来,驱动蛋白作为潜在的肿瘤治疗靶点已成为研究热点。驱动蛋白在癌组织和正常组织中呈差异性表达,可以作为多种肿瘤的诊断标志物;驱动蛋白表达与肿瘤分期相关,可作为独立的危险因素起到预测预后的作用。目前的报道较多地肯定了驱动蛋白在肺癌细胞的增殖、抗凋亡、异常分裂、侵袭转移等过程中的重要作用,但仍存在临床标本量不足、方法局限等问题,且关于驱动蛋白与上下游分子关系的研究较少,参与细胞增殖和凋亡的信号通路等具体分子调控机制仍不十分明确。因此,此后的研究应更多地着眼于驱动蛋白在肺癌发生发展以及侵袭转移中的作用,为肺癌未来的靶向治疗提供依据。
