AEP环化酶的高效原核诱导表达与活性检测体系的建立
2020-01-07胡晓韵何华华彭文舫
胡晓韵,何华华,彭文舫
(湖北大学生命科学学院,省部共建生物催化与酶工程国家重点实验室,湖北 武汉 430062)
0 引言
蛋白酶在自然界含量丰富,种类繁多,在一系列的细胞进程中起着至关重要的作用.绝大多数蛋白酶通过切割底物序列肽键,导致蛋白质的降解,或使之转变为具有生物活性的成熟形式.此外,一些蛋白酶还具有一些特殊功能,例如,天然存在于某些植物中的环化酶,能通过转肽反应将前体肽进行环化,从而形成环肽[1].
2001至2005年,研究者通过对来源于白花蛇舌草[2]、紫罗兰[3]、三色堇[4]和鼠鞭草[5]的cDNA进行克隆,对环化肽前体序列进行了预测和比对.结果显示,其环化结构域的N末端和C末端的加工位点周围有几个氨基酸残基具有高度保守性,如其C末端P1位点的天冬酰胺或天冬氨酸残基.1996年,Philip S. Sheldon等人发现,洋刀豆中凝集素(concanavalin) A在分泌途径的成熟过程与天冬酰胺内切酶有关,该研究证实豆类中存在天冬酰胺内切酶,并进一步发现天冬酰胺内切酶在植物中很常见[6].2014年Giang K T Nguyen等研究发现,来源于蝶豆的天冬酰胺内切酶butelase1可有效环化来自各种生物的非天然肽,证明了天冬酰胺内切酶是天然的环化酶[7].天冬酰胺内切酶识别的底物的C末端加工位点的三肽基序是必需的,通常为Asn-Gly-Leu.
环化肽是一类来源于植物的可环化多肽.在20世纪60年代, Lorents Gran指出,使用白花蛇舌草叶子制成的药茶来诱导分娩并可以促进分娩,后来的研究表明活性成分是植物中一种名为Kalata B1的多肽[8]. Kalata B1是由29个氨基酸组成的环化肽,其具有的6个半胱氨酸残基形成的3个二硫键可以组成一个半胱氨酸结,以增加其结构的稳定性[9].随后,越来越多的环化肽陆续在植物中发现,植物中的环化肽具有杀线虫,杀软体动物,抗HIV,抗神经降压素,收缩子宫,溶血和防污等生物活性[2].
天冬酰胺内切酶(AEPs)催化形成的环化肽具有一个环状主链和一个典型的半胱氨酸结,因其结构异常稳定,所以可作为药物设计框架.然而,从植物组织中不易提取天然AEP环化酶,来源于蝶豆的天冬酰胺内切酶butelase1可有效环化来自各种生物的非天然肽,但该酶在大肠杆菌中进行原核表达未能实现[7].最近,研究者在白花蛇舌草互补DNA文库中鉴定出3种AEP同源蛋白(OaAEP1-3),其中,OaAEP1的一个单氨基酸突变体(Glu371Val,命名为OaAEP1b)可在大肠杆菌中重组表达,其表达量为1.8 mg/L[10].本研究通过克隆白花蛇舌草(Oldenlandiaaffinis)中AEP(OaAEP1b)环化酶编码基因并使其在大肠杆菌中进行表达,在大肠杆菌中利用不同的融合标签,例如麦芽糖结合蛋白(MBP)[11],N-utilization substance A(NusA)[12],小泛素修饰蛋白(SUMO)[13],绿色荧光蛋白(GFP)[14]和泛素(ubiquitin)[15]来促进其可溶性表达和纯化,并验证其对底物肽的切割活性.本研究对AEP环化酶表达量的提高,以及对其酶活性的验证,将为其在肽工程中的应用奠定良好的基础.
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 大肠杆菌XL10-Gold、大肠杆菌BL21(DE3)、载体pET-28a、pRK792-MBP均为本实验室所保存.
1.1.2 试剂 DL 5000 DNA Marker、1 kb DNA Ladder及各种限制性内切酶均购于Takara公司;dNTPs购于Biosharp;Marker Color Prestained Protein Standard购于NEB公司,其余试剂均为国药公司提供.
1.1.3 引物 根据合成的AEP(OaAEP1b)的编码序列(NCBI:ALG36103.1)来设计引物.
1.2 方法
1.2.1 AEP环化酶表达质粒的构建 AEP基因序列合成自生工生物工程(上海)股份有限公司,并作为DNA模板,利用AEP-F/AEP-R(表1)引物对进行扩增,对PCR产物进行切胶回收.用XhoI和NcoI对pET28a载体进行双酶切,切胶回收线性化载体DNA,利用T5核酸外切酶介导的TEDA方法[16]将AEP片段插入到酶切后的pET28a载体中.然后将酶连产物转化到大肠杆菌Gold感受态细胞中,待37 ℃过夜培养后,通过菌落PCR筛选并进行测序验证得到正确的重组质粒pET-28a-AEP.

表1 实验相关引物
在得到AEP环化酶的片段后,利用融合PCR将HRV 3V蛋白酶底物识别序列添加到AEP的N端,然后通过重叠延伸PCR[17]将融合标签和含有HRV 3V底物序列的AEP连接在一起,方法如上所示得到测序正确的重组质粒分别命名为pET-28a-gfp-AEP、pET-28a-MBP-AEP、pET-28a-Sumo-AEP、pET-28a-NusA-AEP和pET-28a-Ubiquitin-AEP.
1.2.2 AEP环化酶底物表达质粒的构建 以基因合成含有Kalata B1-6*His-GST序列的质粒为模板,利用MBP-S-GST-F/ MBP-S-GST--R(表1)引物进行PCR扩增,PCR扩增得到的Kalata B1-6*His-GST片段进行电泳检测并溶液回收;用HindⅢ和NcoI对pRK792-MBP载体进行双酶切,按照1.2.1得到正确的重组质粒命名为pRK792-MBP-Kalata B1-6*His-GST.

将收集的菌体用Lysis Buffer(50 mmol/L Tris-HCl、150 mmol/L NaCl、0.1% triton、1 mmol/L EDTA,pH7.0)进行重悬,并加入Lysozyme混匀后于4 ℃搅拌1 h,搅拌结束后进行超声波破菌(功率35%,每运行3 s停6 s,总共处理10 min),破菌结束后将破菌液在4 ℃,转速17 000 r/min离心30 min,去掉细菌碎片,得到更为完全的破菌上清,随后将破菌上清进行挂柱.
在蛋白纯化柱中加入1 mL Ni-NTA His Bind resin (Takara Blo USA),用Wash Buffer(20 mmol/L bis-Tris、200 mmol/L NaCl、20 mmol/L imidazole,pH7.0)等体积洗涤树脂2次.当蛋白纯化柱中的树脂填充好后,将破菌上清加入到蛋白纯化柱中,静置待树脂沉降下来,用等体积的Wash Buffer来洗涤树脂,反复进行,直至破菌上清全部流经蛋白纯化柱,再用Elution Buffer(20 mmol/L bis-Tris、200 mmol/L NaCl、200 mmol/L imidazole,pH7.0)洗脱目标蛋白.之后,对得到的样品进行SDS-PAGE检测分析,并用Bradford法检测AEP环化酶及其融合蛋白的浓度.同时,用Storage Buffer (5 mmol/L Tris-HCl、100 mmol/L NaCl,pH7.0)对纯化得到的蛋白进行等体积超滤,将纯化得到的环化酶加入到已预冷的超滤管中离心,待液面不再下降后加入Storage Buffer至原始液面高度,反复3次将AEP环化酶的保存Buffer换成Storage Buffer.
1.2.4 AEP环化酶底物融合蛋白在大肠杆菌BL21(DE3)中的表达与纯化 如1.2.3所示,将构建成功的质粒pRK792-MBP -Kalata B1-6*His-GST转化到大肠杆菌BL21(DE3)感受态细胞并进行诱导表达,将收集的菌体用Lysis Buffer(50 mmol/L KH2PO4、300 mmol/L KCl、10 mmol/L imidazole,pH8.0)进行重悬.纯化时用Wash Buffer(50 mmol/L KH2PO4、300 mmol/L KCl、30 mmol/L imidazole,pH8.0)等体积洗涤树脂,在破菌上清全部加入到蛋白纯化柱后,用Elution Buffer(50 mmol/L KH2PO4、300 mmol/L KCl、300 mmol/L imidazole,pH8.0)洗脱目标蛋白.通过SDS-PAGE对纯化得到的样品进行检测,同时用Storage Buffer(5 mmol/L Tris-HCl、100 mmol/L NaCl,pH7.0)将纯化得到的环化酶加入到已预冷的超滤管中离心,待液面不再下降后加入Storage Buffer至原始液面高度,反复3次将AEP环化酶底物融合蛋白的保存Buffer换成Storage Buffer.
1.2.5 利用HRV 3V蛋白酶移除融合蛋白质的融合标签 将融合蛋白Sumo-AEP-6*His和MBP -AEP-6*His与HRV 3V蛋白酶按照摩尔比为80∶1比例混合,并加入10×buffer(50 mmol/L Tris-HCl,150 mmol/L NaCl,5 mmol/L DTT,pH7.5)于4 ℃条件反应过夜以保证融合蛋白被完全切割.反应结束后将整个切割反应体系用Ni柱进行第二次纯化,最后用Elution Buffer洗脱得到AEP环化酶.
1.2.6 AEP环化酶的活性检测 将纯化得到的AEP环化酶在Self-activation Buffer(1 mmol/L EDTA、0.5 mmol/L Tris (2-carboxyethyl) phosphine hydrochloride,用冰醋酸将pH调至4.5)中于37 ℃、pH 4.5的条件下反应5 h.在激活反应过程结束后,将已经激活的AEP环化酶和底物蛋白按照摩尔比为1∶5在Activity Buffer(50 mmol/L sodium acetate、50 mmol/L NaCl、1 mmol/L EDTA、0.5 μmol/L Tris (2-carboxyethyl) phosphine hydrochloride,pH5.0)中进行环化反应,分别于37 ℃水浴锅中反应4、12和24 h.待反应结束后,通过SDS-PAGE检测蛋白质的切割情况.
2 结果
2.1 AEP环化酶基因的原核表达质粒构建以基因合成含有AEP序列的质粒为模板进行PCR扩增从而得到1 356 bp的AEP片段,如图1所示,再进行一轮PCR将HRV 3V蛋白酶的多肽底物识别序列扩增到AEP的N端,然后通过重叠延伸PCR将融合标签和含有HRV 3V底物序列的AEP连接在一起得到含有融合标签的AEP片段.用XhoI和NcoI对pET28a载体进行酶切,如图2所示,按照1.2.1得到正确质粒pET-28a-AEP、pET-28a-gfp-AEP、pET-28a-MBP-AEP、pET-28a -Sumo -AEP、pET-28a-NusA-AEP和pET-28a-Ubiquitin-AEP.

图1 AEP环化酶PCR扩增1和2均为AEP环化酶的PCR产物;

图2 pET-28a载体酶切图1:用NcoI和XhoI双酶切的pET-28 a载体;2:未酶切的pET-28a载体;
2.2 AEP环化酶的表达与纯化将AEP环化酶及其融合蛋白按照1.2.3中的方法表达并纯化如图3所示,表达的融合蛋白由四部分组成:融合蛋白标签、HRV 3V蛋白酶识别序列、用于亲和纯化的6*His标签和AEP环化酶.融合表达蛋白质1(GFP+AEP)大小为:76.0 kD,融合表达蛋白质2(SUMO+AEP)大小为:61.0 kD,融合表达蛋白质3(MBP-AEP)大小为:92.0 kD,融合表达蛋白质4(NusA-AEP)大小为:104.0 kD,融合表达蛋白质5(Ubiquitin+AEP)大小为:58.0 kD.结果显示:表达的融合蛋白质与预测的大小一致,经质谱分析确定在大肠杆菌BL21(DE3)中所表达的蛋白质为AEP环化酶,表明不同的融合蛋白质在大肠杆菌BL21(DE3)中均有表达,并且可以通过Ni柱亲和层析可以纯化得到表达的融合蛋白,其中使用NusA、GFP和Ubiquitin作为融合标签时,对AEP环化酶的表达没有明显的促进作用,而SUMO和MBP可以明显促进AEP环化酶的表达.
2.3 利用HRV 3V蛋白酶移除融合蛋白的标签融合标签可促进目的蛋白的可溶性表达,但在验证目的蛋白生物活性时,为了避免额外的氨基酸序列对目的蛋白相关特性的影响,往往需要对融合标签进行去除.本研究中,我们在融合标签和AEP环化酶之间加入了HRV 3V蛋白酶的底物识别位点,因此,可以按照1.2.5利用HRV 3V切割MBP-AEP-6*His融合蛋白和SUMO-AEP-6*His融合蛋白中底物序列LEVLFQ/GP来分离融合标签和AEP环化酶.反应结束后将整个切割反应体系用Ni柱进行第二次纯化.因AEP环化酶的C端含有6*His标签,故可以用来纯化AEP环化酶,从而将融合标签蛋白和AEP进行有效的分离,得到纯AEP环化酶.将Elution Buffer洗脱下来的蛋白质用SDS-PAGE检测,结果显示(图4),SDS-PAGE中AEP条带的大小与预测的49.7 kD大小一致,由此分别成功得到了去掉融合标签MBP和SUMO的纯的AEP环化酶,并根据Bradford方法测其蛋白质浓度,其蛋白浓度为分别为(3.6±0.05) mg/L和(2.8±0.05) mg/L.

图3 不同融合蛋白纯化的SDS-PAGE分析1:AEP-6*His; 2:GFP-AEP-6*His; 3:NusA-AEP-6*His;4:Ubiquitiniquitin-AEP-6*His;5:SUMO -AEP-6*His;6:MBP -AEP-6*His;M:蛋白预染
图4 移除融合标签的AEP环化酶的SDS-PAGE分析M:蛋白预染Marker;1:移除融合标签MBP后的AEP环化;2:移除融合标签SUMO后的AEP环化

图5 AEP环化酶底物融合蛋白纯化的SDS-PAGE分析 1:第一次用Elution buffer洗脱的 AEP环化酶底物融合蛋白;2:第二次 用Elution buffer洗脱的AEP环化酶 底物融合蛋白;M:蛋白预染
2.4 AEP环化酶底物的设计、表达与纯化由于AEP天然底物Kalata B1序列较短,为了方便检测AEP对其切割活性,研究中我们在底物 Kalata B1不变的基础上,在两端分别加上融合蛋白MBP和GST,倘若发生切割作用,底物融合蛋白会有大约26.0 kD的大小变化,这样也就可以确定AEP环化酶是否有活性,按照1.2.4对AEP环化酶底物融合蛋白进行表达纯化,如图5所示,AEP环化酶底物融合蛋白的大小为74 kD,SDS-PAGE中条带的大小与预测的大小一致,因此我们得到了底物融合蛋白质,并进行了较好的纯化.
2.5 AEP环化酶的活性检测Kalata B1的前体结构经验证是具有可折叠的环化结构域,可以用来作为AEP环化酶的底物.AEP环化酶识别其C末端的天冬酰胺(N)-甘氨酸(G)-亮氨酸(L) 3个氨基酸,并在天冬酰胺和甘氨酸之间发生切割,C端切割后暴露的-COOH与自由存在的N端-NH3进行连接并发生环化反应,使线性化的多肽底物成为一个环肽,本研究在底物Kalata B1的两端分别加上融合蛋白MBP和6*His-GST,因AEP环化酶作用在位于底物多肽C端的氨基酸N和G之间,因此当AEP环化酶与融合底物作用后,通过比较融合蛋白的大小变化即可判断AEP环化酶是否可以在这种形式的融合蛋白质中起作用.

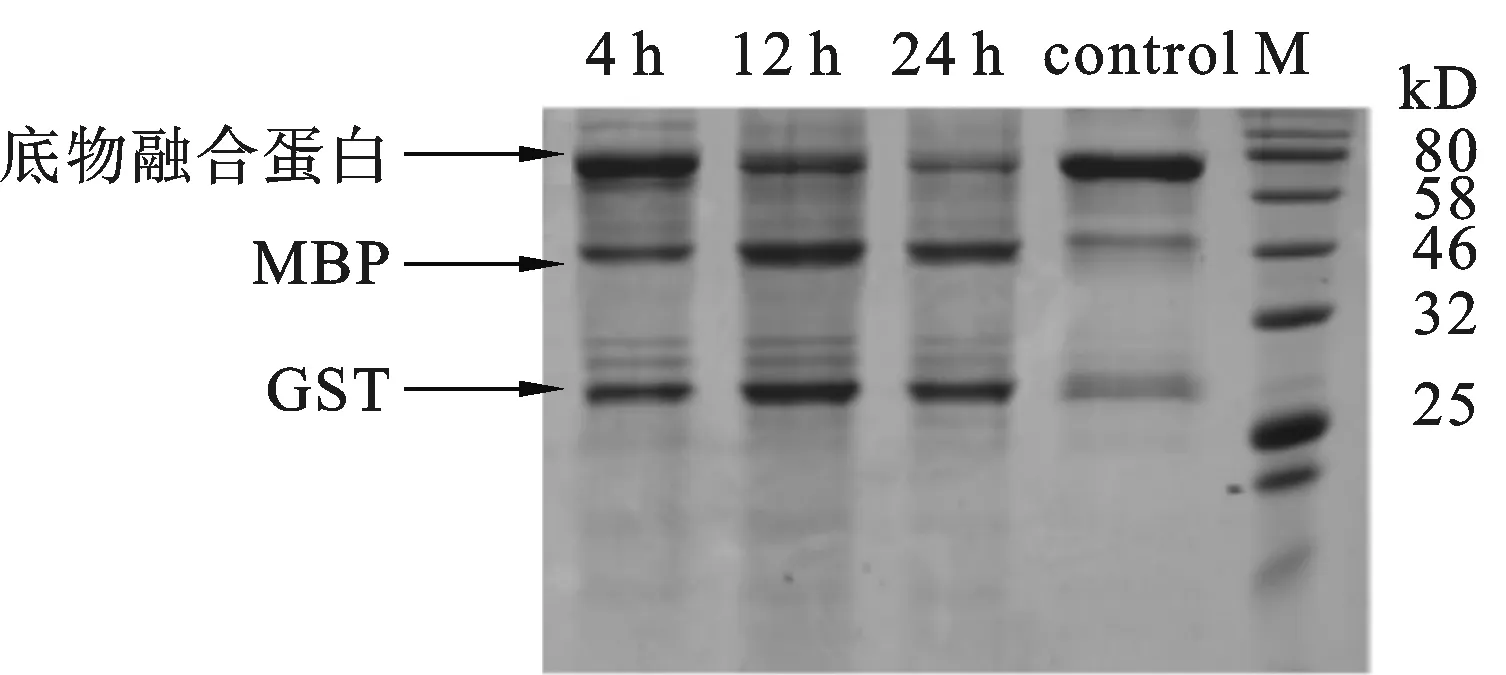
将移除融合标签MBP后纯化得到的AEP环化酶在激活缓冲液中于37 ℃,pH 4.5的条件下反应5 h,在激活反应过程结束后,将已经激活的AEP环化酶和底物融合蛋白按照摩尔比1∶5在反应缓冲液中进行反应,于37 ℃水浴锅中做不同时间切割反应: 4、12和24 h,并在反应结束后进行SDS-PAGE检测底物融合蛋白切割情况.如图6所示,AEP环化酶对Kalata B1的C末端有切割作用,根据Image J定量计算显示,在反应4 h时AEP环化酶可切割16.5%的底物蛋白,在反应12 h时AEP环化酶可切割44%的底物蛋白,在反应24 h时AEP环化酶可切割66.2%的底物蛋白.AEP环化酶底物融合蛋白的大小为74 kD,在与AEP环化酶反应后,底物融合蛋白会被切割为大小分别为46.3 kD的MBP-N端Kalata B1,和28.1 kD的GST-C端Kalata B1.由于环化结构域的线性状态大小为3.2 kD,因分子量太小,无法在SDS-PAGE上看到蛋白质条带.
3 讨论
天冬酰胺内切酶(AEP)是一种半胱氨酸蛋白酶,在植物中分布广泛,现在已经证明了天冬酰胺内切酶是天然的环化酶,AEP在低pH条件下移除N-末端和C-末端前结构域而被加工成其成熟形式[18],其催化形成的环化肽是来源于植物的可环化多肽,在结构上除了它们的环状主链外,还具有由其6个保守的半胱氨酸残基形成的3个二硫键的半胱氨酸结,这种结构特征的组合使得它们异常稳定,因此可作为药物设计应用的潜在支架或蛋白质工程框架.
OaAEP1b是从产生环化肽的植物白花蛇舌草的基因组中鉴定出的AEP同种型,与其他天冬酰胺内切酶(AEP)具有相似的结构折叠.本研究通过在大肠杆菌中利用不同的融合标签促进AEP环化酶的可溶性表达,之后通过移除融合标签得到表达量较高的AEP环化酶,其表达量为(3.6±0.05) mg/L,高于原先文献报道的1.8 mg/L;对于AEP环化酶的活性验证,AEP环化酶的活化过程应该分为两个部分,先切割后环化,本研究证明了其可对kalata B1前体蛋白的C末端进行切割,在之后的实验中将通过构建一种串联底物来验证环化活性,由于SUMO蛋白质大小为11.0 kD,除了能够较好地在SDS-PAGE中观察到并且具备环化的可能性,选取SUMO作为环化底物,在其两端分别加上较长的linker和AEP环化酶的识别序列,并在C端的较长的linker和AEP环化酶的识别序列之间加入TEV蛋白酶的底物序列,在与AEP环化酶作用后利用TEV蛋白酶对反应后底物进行切割,根据切割后底物大小的变化或者条带数量来鉴定是否环化,并定量分析从而确定环化效率.在进一步确定AEP环化酶的环化活性后,利用AEP环化酶对人工设计的活性肽进行环化,对于提高其稳定性和延长其半衰期具有极其重要的应用意义.
