Development of a method to quantify total and free irinotecan and 7-ethyl-10-hydroxycamptothecin(SN-38)for pharmacokinetic and bio-distribution studies after administration of irinotecan liposomal formulation
2020-01-06YongjunWng
,Yongjun Wng,
aWuya College of Innovation,Shenyang Pharmaceutical University,Shenyang 110016,China
bKey Laboratory of Structure-Based Drugs Design&Discovery of Ministry of Education,Shenyang Pharmaceutical University,Shenyang 110016,China
ABSTRACT In 2015,liposomal formulation of irinotecan(ONIVYDE)has been approved by FDA and widely applied in the treatment of pancreatic cancer.ONIVYDE is a novel liposome formulation,entrapping CPT-11 in the aqueous core of vesicles using a modified gradient loading method.Due to toxicity concerns,it is essential to explore a rapid and reliable method to effectively isolate and quantify the non-liposomal,namely,free CPT-11and total CPT-11 in plasma.This study focuses on separation of non-liposomal CPT-11,evaluation of the pharmacokinetics of free CPT-11 and total CPT-11 and bio-distribution after intravenous administration of CPT-11 liposome.Free CPT-11 in plasma was separated by solid-phase extraction(SPE).The amount of total CPT-11 and main metabolite 7-ethyl-10-hydroxycamptothecin(SN-38)in plasma was quantified by ultra-performance liquid chromatography-MS/MS.The calibration curves fitted well and lower limit of quantitation for SN-38,free CPT-11,total CPT-11 and CPT-11 in tissue and were 5 ng/ml,10 ng/ml,4.44 ng/ml and 25 ng/ml respectively.The recoveries,precision and accuracy of the method appear satisfactory.Using this method,the pharmacokinetics and bio-distribution of CPT-11 liposome formulation after an intravenous dose of 2.5 mg/kg were then investigated.
Keywords:Irinotecan Liposome UPLC-MS-MS SPE Pharmacokinetics Bio-distribution
1.Introduction
Nanoparticles like small unilamellar liposome have shown their promise for improving the pharmacokinetics and tumor localization of encapsulated anticancer drugs[1].In 2015,FDA approved ONIVYDE(irinotecan liposome injection)in combination with fluorouracil/folinic acid based on the result of clinical study.Compared with fluorouracil/folinic acid,this dosage regimen significantly pro-longed overall survival of patients with advanced and metastatic pancreatic cancer[2].ONIVYDE was prepared by a modified active loading method,was able to entrap irinotecan(CPT-11)at an extreme high drug to lipid ratio,retain encapsulated CPT-11 stable in vesicles,and also protect CPT-11 from hydrolysis and metabolic conversion to 7-ethyl-10-hydroxycamptothecin(SN-38)during the systemic circulation[3].For CPT-11 is an ionizable drug,loaded by active loading method,drug release rate and circulation half-life determined the therapeutic activity of liposomal drug delivery systems.And only if liposomes retain CPT-11 over several hours to days will achieve high drug accumulation in tumor[4].Therefore,it is important to control the ratio of free drug,monitor the pharmacokinetics(PK)and tissue distribution properties to fully understand and the therapeutic efficacy and side effects of liposome formulation.
To get a clear relationship of pharmacokinetics between total drug and non-liposomal drug,namely the free drug,there is an insistent demand of a sophisticated analytical technique to separate free CPT-11 from liposome in plasma.Meanwhile,the developed separation method should have a consistent recovery and extraction efficiency of free CPT-11.Moreover,formulation integrity should be maintained without leakage or breaking of the liposome during sample preparation and storage.Up to now,there are several methods have been reported to achieve this end for vincristine liposome,amphotericin B liposome and doxorubicin liposome,such as solid-phase extraction(SPE)[5-10],ultrafiltration[11],ion-exchange chromatography[12],and capillary electrophoresis[13,14].Nevertheless,no method has been observed for the separation and detection of free CPT-11 and liposomal CPT-11.Furthermore,as high risk of liposome leakage using ultrafiltration and high sample dilution during gel chromatography,these methods have obvious limitations in applying for CPT-11 liposome determination from biological samples.
Based on previous reports[5-10,15],SPE is the most feasible method for separation of non-liposomal CPT-11 and liposomeencapsulated CPT-11 due to better security and relative rapidness.However,the separation and quantification of nonliposomal,liposomal CPT-11 and metabolite SN-38 was still challenging because carboxylesterase in rat plasma is highly active.Considering that the catalysis of carboxylesterase is related to protein concentration,pH,ionic strength,and these conditions are easily changed under SPE processing[16,17],the working process especially the solution involved in SPE should be controlled properly.In the present study,we build a method to(i)separate free CPT-11(F-CPT-11)from liposome by SPE without damaging liposome(ii)achieve total release of encapsulated CPT-11 and extract total CPT-11(T-CPT-11)from liposome using protein precipitation extraction(iii)detect CPT-11 in liver(L-CPT-11),heart,spleen,lung and kidney(OT-CPT-11)of mouse using protein precipitation extraction.And the developed method had been proved to be valid for the separation and quantitation of non-liposomal drug and total drug in plasma and tissue samples according to the FDA guidelines on bioanlytical method[18],allowing the study of pharmacokinetics and bio-distribution after administration of CPT-11 loaded liposome.
2.Materials and methods
2.1.Chemicals reagents
Irinotecan was kindly supported by Jiangsu Hengrui Medicine Co.,Ltd.(China).Sucrose octasulfate was obtained from Wuhan Hengruikang Reagent Co.,Ltd.(China).Methanol,acetonitrile and dichloromethane(HPLC grade)was purchased from Tianjin Kemer Chemical Reagent Co.,Ltd.(Tianjin,China).Disaturated phosphatidylcholine(DSPS),cholesterol(chol)and 2-Distearoyl-snglycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000](mPEG2000-DSPE)were obtained from Shanghai Advanced Vehicle Technology L.T.D.Co.Sephadex G-75 size exclusion resins were procured from Sigma Chemical Company(St Louis,USA).All other reagents and solvents mentioned were of analytical grade.
2.2.Instrumentations and UPLC-MS/MS conditions
Mass spectrometric detection was conducted in a Xevo-TQS triple quadrupole mass spectrometer(Waters,Milford,MA,USA)equipped with an electrospray ionization(ESI)source in the positive mode.And the quantification of analytes were operated in the multiple reaction monitoring(MRM)mode.Camptothecin(CPT)which has similar physicochemical properties as CPT-11,was selected as internal standard(IS).The MS operational conditions were optimized,MRM transitions of SN-38,CPT-11and IS were m/z 393.2→349,m/z 587.4→167.1 and m/z 349.2→249.0 respectively.Capillary voltage was 3.5 kV.And cone voltage was 30 V for both IS and CPT-11,and 35 V for SN-38.The collision energies of CPT-11,SN-38 and the IS were 37,34 and 35 eV,respectively.
LC separations were performed on an ACQUITY UPLC system(Waters Corp.,Milford,MA,USA)with UPLC BEH C18 column(2.1 mm×50 mm,1.7 μm;Waters Co.,Ltd.,USA).Acetonitrile(A)and water containing 0.1%formic acid(B)were used as the mobile phase,and the elution was performed by 27%(A):73%(B)(v/v)at a flow rate of 0.3 ml/min.The injection volume was controlled at 20 μl.The column temperature and the auto sampler temperature was set to 60°C and 4°C respectively.
MassLynxTMVersion 4.1 software was applied for data acquisition and analysis.
2.3.Preparation of calibration standards and quality control(QC)
Stock solutions of CPT-11(500 μg/ml),IS(500 μg/ml)and SN-38(500 μg/ml)were prepared by dissolving weighed corresponding analytes into volumetric flasks and stored at-20°C before analyze.CPT-11,IS were dissolved in methanol,and SN-38 was dissolved in dimethyl sulfoxide.To provide intermediate working solutions,the stock solutions were further diluted with methanol/water mixture(50:50,v/v).The calibration standard for all plasma samples were prepared at 7 concentrations,ranging from 10 ng/ml to 10 000 ng/ml for FCPT-11,from 4.4 ng/ml to 20 000 ng/ml for T-CPT-11 and from 5 ng/ml to 1000 ng/ml for SN-38.Corresponding QC concentrations were 10,50,500,8000 ng/ml for F-CPT-11,4.4,11.1,1778,8888 ng/ml for T-CPT-11,and 5,10,200,800 ng/ml for SN-38.Liver QC samples and other tissue QC samples were prepared at 25,62.5,500 and 800 ng/ml,and corresponding calibration standards were prepared ranging from 25 to 1000 ng/ml.
2.4.Sample preparation
2.4.1.Plasma sample preparation for CPT-11 and SN-38
Part A:SPE for F-CPT-11
F-CPT-11 in plasma was separated from CPT-1 1 liposome using SPE with an Oasis HLB column(1 cc/30 mg,Waters Corp.,Milford,Massachusetts,USA)connected with a vacuum air pump.Previous study has shown that chilling samples at 4°C could probably sustain the stability of lactone form[19].Thus,all solvent involved in SPE process was maintained at 4°C.Firstly,SPE cartridge was preconditioned with 1.0 ml methanol and 1.0 ml water.Then,75 μl 5% glucose solution were added to 50 μl CPT-11 spiked plasma,the mixture were passed through cartridges without vacuum.Then,20 μl ZnSO4,75 μl 5%glucose solution and 50 μl IS working solution(5 μg/ml)were added to 50 μl plasma.Samples were mixed by vortex for 1 min and then centrifuged at 7500 rpm for 3 min.Supernatants were then collected,and passed through the conditioned cartridges with vacuum.After complete passage of samples,each cartridge was further rinsed with 2 ml of PBS,1 ml of 3.75% ammonia.Finally,each cartridge was washed with 1 ml methanol containing 2% acetic acid,target analyte in the rinsed cartridge was collected and evaporated under a stream of nitrogen at 37°C.Residues were then dissolved with 200 μl methanol/water containing 0.1%acetic acid(70:30,v/v)and centrifuged at 13,000 rpm for 5 min.20 μl of the supernatant were injected into the UPLC separation system.
Part B:Protein precipitation extraction for T-CPT-11 and SN-38
Fifty microliter of IS solution(5 μg/ml in methanol)was firstly added to 50 μl plasma(QC samples and calibration standards were spiked with corresponding working solutions)and vortex mixed for 30 s.Then,400 μl of methanol containing 0.1% acetic acid was added to the plasma samples,after mixing by vortex for 5 min,the mixture was centrifuged at 15 000 rpm for 10 min.Finally,a 20 μl sample of supernatant were injected into the UPLC column for analysis.
2.4.2.Tissue sample preparation for CPT-11
Firstly,50 μl IS solution(5 μg/ml in methanol)and 850 μl of methanol/acetonitrile(50:50,v/v)were added to 50 μl tissue samples(QC samples and calibration standards were spiked with corresponding working solutions).Samples were then mixed by vortex for 1 min,and centrifuged at 15,000 rpm for 10 min to precipitate possible particulate matter,and the supernatant was collected and then evaporated under a stream of nitrogen at 37°C.Residues were then reconstituted in 100 μl of methanol/water containing 0.1%acetic acid(70:30,v/v)and centrifuged at 13 000 rpm for 5 min.Finally,a 20 μl sample of supernatant were injected into the UPLC column for analysis.
2.5.Method feasibility
It is necessary to assess the feasibility of SPE method before the method validation,and prove that the separation and quantitation method could be used for pharmaceutical and bio-distribution study.Moreover,the sample preparation methods(both SPE and protein precipitation extraction)must be appropriate for the separation of F-CPT-11 from liposome.Meanwhile,liposome integrity must be ensured.In this research,method feasibility was performed by determining the part of F-CPT-11(F %) and T-CPT-11(T%)in QC samples(QC samples were spiked with CPT-11 liposomes).And F-CPT-11(F %)was calculated by the formula:F %=CF/CN×100,here CFis the F-CPT-11 concentration detected in QC samples and CNis the nominal concentration of the CPT-11 added in QC samples.And CFmay compose of originally present F-CPT-11 in liposome formulations as well as the part of released CPT-11 during mixing and SPE separation process.If SPE method could separate F-CPT-11 from liposome,the value of F1% after the first SPE process should equal to the encapsulation rate.To further prove the liposome integrity throughout the SPE process,liposome fraction in the first SPE process were collected.The collected liposome fraction was then re-run through a new preconditioned cartridge.Theoretically,if the liposomes remain intact and the CPT-11 was not released from the collected liposome fraction during the second SPE process,the value of F2% after the second SPE process should equal to zero.
Moreover,the value of T-CPT-11(T%)could be used to verify the feasibility of protein precipitation extraction process.In the equation:T%=CT/CN×100,CTis the T-CPT-11 concentration detected in QC samples,and CNis the nominal concentration of the T-CPT-11 added in QC samples.If protein precipitation process could deposit protein completely and ensure a total release of drug molecular from liposomes,the value of T%should be within 85%-115%.
2.6.Method validation
In this study,analytical methodology of analytes were built for pharmacokinetics studies of CPT-11 liposome.As liver plays an essential role in drug metabolism and excretion when doing bio-distribution studies,so we build analytical methodology for liver and mixture of other tissues(heart,spleen,lung,kidney)respectively.Moreover,selectivity,linearity of calibration curves,lower limit of quantitation(LLOQ),precision and accuracy,recovery and matrix effect and stability of the analytical methodology were validated.And analytical methodology was run on three consecutive days.
2.6.1.Specificity
Specificity of the methodology was evaluated by comparing the chromatograms of blank plasma,blank liver homogenates,and homogenates mixture of blank tissue(heart,spleen,lung and kidney)with the corresponding analytes spiked plasma or tissue homogenates.
2.6.2.Linearity of calibration curves and LLOQ
Calibration curves of plasma samples were prepared by diluting corresponding stock solutions to seven final concentrations,ranging from 5 ng/ml to 2000 ng/ml for SN-38,10 ng/ml to 10 000 ng/ml for F-CPT-11,and 4.4 ng/ml to 20 000 ng/ml for T-CPT-11.As for tissue samples,calibration curves achieved by using seven nonzero liver homogenates and seven nonzero tissue homogenates calibration standards from 25 ng/ml to 1000 ng/ml for CPT-11.Calibration curves linearity was analyzed by plotting peak area ratios of CPT-11 and SN-38 versus the nominal concentration.Moreover,least-square regression using a 1/(concentration)2weighting was applied to evaluate linearity relationship.The correlation coefficients(r2)needed to be above 0.99.The LLOQ of the calibration curve was defined by using six lots of blank plasma samples and blank tissue homogenates.Both precision and accuracy are required to be less than 20%.
2.6.3.Precision and accuracy
For all analytes,intra-day precision,inter-day precision and accuracy were estimated by analyzing QC(low,middle and high)samples on three separated days in six replicates.The precision and accuracy was defined as the relative standard deviation(RSD%)and the relative error(RE%).Moreover,the acceptability for precision was within 15%RSD and that of accuracy was within 15%RE of the theoretical values.
2.6.4.Extraction recovery and matrix effect
The extraction recovery of CPT-11 and SN-38 were analyzed by comparing the peak areas of the extracted QC(low,middle and high)samples with average peak area of CPT-11/SN-38 added blank samples after extraction.Moreover,at each QC concentration six replicates were determined.And matrix effect was determined as the ratio of the peak area from extracted with blank plasma/tissue homogenates to the mean peak area from extracted without plasma/tissue homogenates at QC concentrations.
2.6.5.Stability
Stability of plasma samples and tissue samples were tested including auto-sampler storage,freeze-thaw cycles,and shortterm,long-term storage stability at three different levels(QC samples).The auto-sampler stability was evaluated by placing the extracted QC samples in the auto-sampler,and keeping them at 4°C for 24 h.Freeze-thaw stability was evaluated by comparing peak areas of QC samples after 3 cycles of freeze-thaw(-20°C to ambient temperature)with peak areas of newly prepared QC samples.Short-term stability of FCPT-11was invested by keeping the QC samples at 4°C for 6 h,and that of T-CPT-11,L-CPT-11,OT-CPT-11 and SN-38 was conducted at 25°C for 12 h for.And the long-term stability was evaluated by comparing the concentration of newly prepared QC samples with that of QC samples stored at-80°C for 1 months.Moreover,robustness study of the developed method against small variations of column temperature(from 59°C to 61°C)was also conducted.
2.6.6.Dilution effect
Stock solutions of CPT-11(120 μg/ml)and SN-38(8 μg/ml)were prepared by dissolving weighed corresponding analytes into volumetric flasks.Then,CPT-11 stock solution and SN-38 stock solutions were further diluted with blank plasma for 20-fold and 10-fold(v/v),respectively.CPT-11/SN-38 were then extracted as protocol in 2.4.1
2.7.Preparation and characterization of CPT-11 liposome
2.7.1.Preparation of CPT-11 liposome formulation
First,solution of triethylammonium salt sucrose octasulfate(TEA8SOS)was prepared.The concentration was adjusted to 0.65 mol/l,and the pH was 5.5-6.0.Then,the mixture of DSPC(6.81 mg/ml),chol(2.22 mg/ml)and mPEG2000-DSPE(0.12 mg/ml)were combined in 50%(w/v)ethanoic solution and then mixed with TEA8SOS solution at 65°C for 30 min.The lipid suspension was then extruded 20 times at 65 °C.Unentrapped TEA8SOS were then removed by Sephadex G-75 gel filtration chromatography,and the gel filtration was eluted with pH 6.5 HEPES/NaCl(17/144,mM/mM)buffer.After buffer exchange,CPT-11 HCl was then added to liposome at 4.3 mg/ml.The solution was then incubated at 65±5°C for 1 h and immediately chilled on ice for 20 min.
2.7.2.Characterization of CPT-11 liposome formulation
The average size and zeta potential of prepared liposomes was analyzed by dynamic light scattering(Zetasizer Nano ZS90;Malvern Instruments Ltd.,Malvern,UK).Each sample was measured in triplicate for 14 cycles at 25°C.Then,ultrafiltration centrifugal method was used to assess the entrapment efficiency of liposome.In this method,100 μl liposome was added into the ultrafiltration concentrator(10 kDa)and then centrifuged(3000 rpm)for 30 min to make sure the unentrapped CPT-11 follow to the bottom,and the content was the part of the entrapped liposome.The content of F-CPT-11 and liposomal fraction of CPT-11 were then determined by high-performance liquid chromatography(HPLC)on a reverse Agilent 5 TC-C18(2)column(250 mm×4.6 mm,4.5 μm).The entrapment efficiency was then calculated by the equation:entrapment efficiency %=liposome fraction of CPT-11/(liposome fraction of CPT-11+F-CPT-11)∗100.
2.8.Pharmacokinetic analysis and bio-distribution study
The developed method was finally applied in the quantification of SN-38,F-CPT-11 and T-CPT-11 for pharmacokinetic study in rat plasma and bio-distribution study(including heart,liver,spleen,lung and kidney)in mice.Animal experiments involved were performed according to the“Guidelines for the Care and Use of Laboratory Animals”,and were approved by the Animal Ethics Committee of Shenyang Pharmaceutical University(Shenyang,China).
Each rat was given at 2.5 mg/kg CPT-11 liposome by intravenous administration.Then,at 0.083,0.25,0.5,1,2,4,6,8,12,and 24 h,0.2-0.3 ml blood samples was obtained through orbital venous plexus and collected in polyethylene tubes.Collected blood samples was immediately centrifuged at 10 000 rpm for 5 min,upper plasma was then stored at-80°C until analysis.
In bio-distribution study,each mouse(22±2 g)was given 2.5 mg/kg CPT-11 liposome formulation by intravenous administration.Vital organs(heart,liver,spleen,lung and kidney)were then removed at 6 h,24 h and 48 h and washed with saline thoroughly to remove blood.Then,tissues were dried,weighed and diluted 1:1 by saline and homogenized with saline carefully in an ice bath and stored at-80°C until analysis.
In this study,the DAS 2.1.1 was used for pharmacokinetic parameters analysis.All values were expressed as mean±SD.
3.Results and discussion
3.1.Method optimization
As carboxylesterase is in a high active state in rat plasma,inactivating carboxylesterase and preventing the transformation of CPT-11 into metabolite SN-38 during sample processing was a major concern.The lactone forms CPT-11 and SN-38 have a closedα-hydroxy-δ-lactone ring,which play a key role in the interaction between CPT-11/SN-38 with topoisomerase I[16].However,it should be noted that this functional lactone ring was liable and probably hydrolyzed to the carboxylate form[20].The hydrolysis of the lactone ring occurred under a pH-dependent equilibrium,and the rate is dependent on pH[17],ionic strength[16],and protein concentration[17].In the current study,liposome could stabilize entrapped CPT-11 as active lactone form.And it was also possible to ensure that CPT-11 and SN-38 were quantified as total lactone form by adjusting the pH during pre-treatment and chromatographic separation process[21].
The SPE separation process of F-CPT-11 has to be achieved without breaking the liposome integrity.Previous study has shown that chilling samples at 4°C could probably sustain the stability of lactone form[19].Thus,all solvent involved in SPE process was maintained at 4°C.Moreover,zinc sulfate which has strong capacity of enzyme inactivation and compatibility of mass analyses was added to plasma to inactivate carboxylesterase[22,23].Mass spectroscopy parameters were optimized in positive ion mode to achieve sensitivity and selectivity.CPT-11,IS and SN-38 spectrometric analysis were scanned in ESI positive,and MRM mode was used for signal acquisition.It was found that CPT-11,IS and SN-38 were more sensitive in positive ion mode,were able to accept proton and generate[M+H]+ions.And the base peak of CPT-11,IS and SN-38(Fig.1)was selected at m/z 587.4,393.2 and 349.2 respectively.In addition,collision energy was further optimized to acquire more abundant product ions of three analytes.The most prominent and stable fragments of CPT-11,SN-38 and IS were selected at m/z 167.1,349.2 and 305.1 at a collision energy of 37 eV,34 eV and 35 eV respectively.And the source temperature was set at 120°C.
3.2.Method validation
3.2.1.Specificity
The specificity of SPE method and protein precipitation extraction were evaluated by analyzing six different batches of blank plasma/tissue versus the corresponding analytes spiked plasma/tissue.The typical chromatograms are shown in Fig.2 and Fig.S1.The chromatographs demonstrate the sensitivity,selectivity and specificity of both methods.Retention time of 0.67,1.63,and 1.28 min were observed for CPT-11,IS and SN-38 respectively.Moreover,there was no obvious interference from blank plasma and blank tissue at corresponding retention times.
3.2.2.Linearity calibration curves and LLOQ
The current assay offered LLOQ of 10 ng for quantitation of F-CPT-11,4.44 ng for T-CPT-11,25 ng for L-CPT-11 and OT-CPT-11,and 5 ng for SN-38.These limit of quantitation were sufficient enough for further studies.And these standard calibration curves exhibited excellent linearity in the linear range.The typical regression equations were y=9.04×10-3x+4.32×10-3(r2=0.99)for F-CPT-11,y=1.05×10-3x-2.77×10-2(r2=0.99)for TCPT-11,y=2.55×10-3x-3.08×10-2(r2=0.99)for L-CPT-11,y=3.05×10-3x+6.51×10-2(r2=0.99)for OT-CPT-11,and y=2.57×10-3x-7.58×10-3(r2=0.99)for SN-38.
3.2.3.Precision and accuracy
The intra-day and inter-day precision and accuracy of three analytes were validated by analyzing LLOQ(n=6)and QC samples(n=6,)at three levels in plasma and tissue.As shown in Table 1,the established method was accurate and repeatable,tested samples were within the acceptable criteria(RSD%<15%).
3.2.4.Recovery and matrix effect
SPE and protein precipitation extraction were applied in this study for extracting F-CPT-11 and T-CPT-11 from plasma and tissue.The mean extraction recoveries and matrix effect for two methods were shown in Table 2.Considering that the extraction recoveries of tissue samples were still reproducible and compatible,the developed method remain sufficient within the detection limit.Moreover,as the matrix effect was also within the acceptable range(below 115%),reproducible and did not alter the precious or accuracy.
3.2.5.Stability
The stability of CPT-11 and SN-38 in plasma and tissue was investigated under a variety of conditions:short-term stability,long-term stability,freeze-thaw stability and auto sampler stability.The results were shown in Table S1.Here,the shortterm stability of the F-CPT-11 samples was investigated at 4°C to reduce likely occurrence of liposome leakage.As shown,the results indicated that F-CPT-11 was stable at 4°C for 6 h and at-30°C for 2 weeks.Other samples were stable under analysis,all RE values were less than 15%.The stability of this UPLC-MS/MS method against column temperature were also evaluated.As shown in Table S2,tested samples were within the acceptable criteria(all RE and RSD values were less than 15%).Moreover,RSD for inter-and intra-batch were 12.32%and 5.04%respectively,all less than 15%.The results indicated that CPT-11 was stable for against small variations of column temperature.
3.2.6.Dilution effect
To ensure the accuracy of quantitation,plasma samples with high concentration of T-CPT-11/SN-38 must be diluted before processing.Therefore,the dilution effect of T-CPT-11 and SN-38 in rat plasma were measured.As shown in Table S1,the established dilution method was accurate and repeatable,tested samples were within the acceptable criteria(RSD%<15%,RE%<15%).
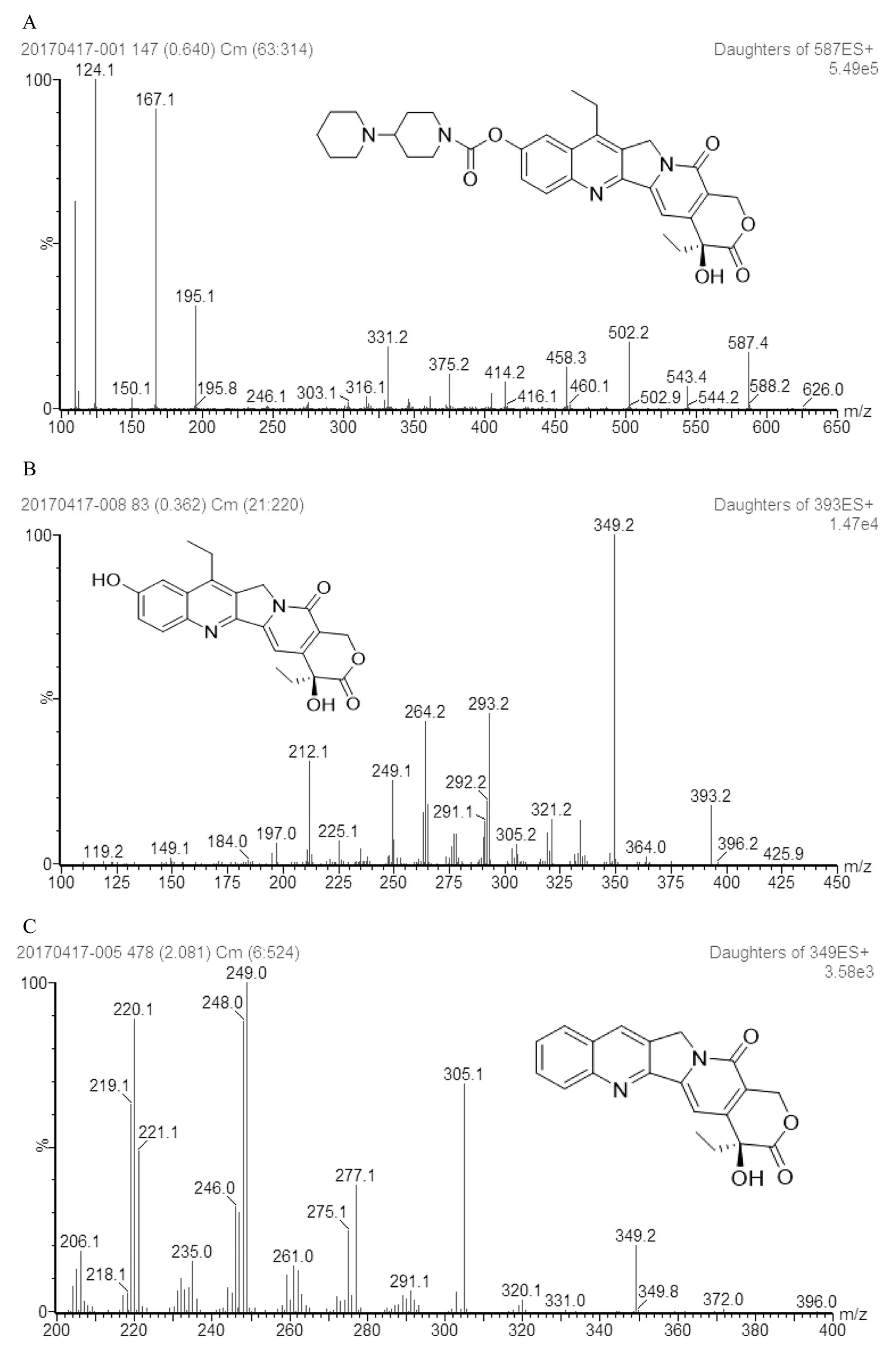
Fig.1.-Product ion mass spectra of CPT-11(A),SN-38(B),and CPT(C).

Fig.2.-Typical UPLC chromatograms of blank plasma samples,blank tissue samples(A),UPLC chromatograms of plasma samples and blank tissue samples spiked with the IS(B),UPLC chromatograms of CPT-11 in plasma samples from a rat at 1 h and tissue sample from a mice at 6 h after intravenous administration of CPT-11 liposome(C),UPLC chromatograms of IS and SN-38 in plasma samples from a rat at 1 h after intravenous administration of CPT-11 liposome(D).
3.3.Method feasibility
Protocol for separating F-CPT-11 from plasma was shown in Fig.3.And the feasibility results for both SPE and protein precipitation extraction were summarized in Table 3.To quantify the released F-CPT-11 during the extraction process,QC samples spiked with CPT-11 liposome solution(n=3)were extracted at three concentrations by using SPE.After one-step extraction,F-CPT-11(F1%)in LOQ,MOQ,and UOQ were 0,4.90% and 1.59% respectively.And the corresponding F2%value in three QC levels were 0%,0.92%,1.34% respectively.Compared with the amount of F-CPT-11 originally present in CPT-11 liposome solution,the value of F1% and F2% in QC samples were below than 5% and negligible.Thus,this result showed that SPE method could separate the F-CPT-11 from liposomes without changing its percentage and causing liposome damage during the extraction.And the concentrations of three QC samples spiked with liposomal CPT-11 were determined after extraction using protein precipitation,indicating that the protein precipitation extraction could achieve complete release of encapsulated CPT-11 from liposome.
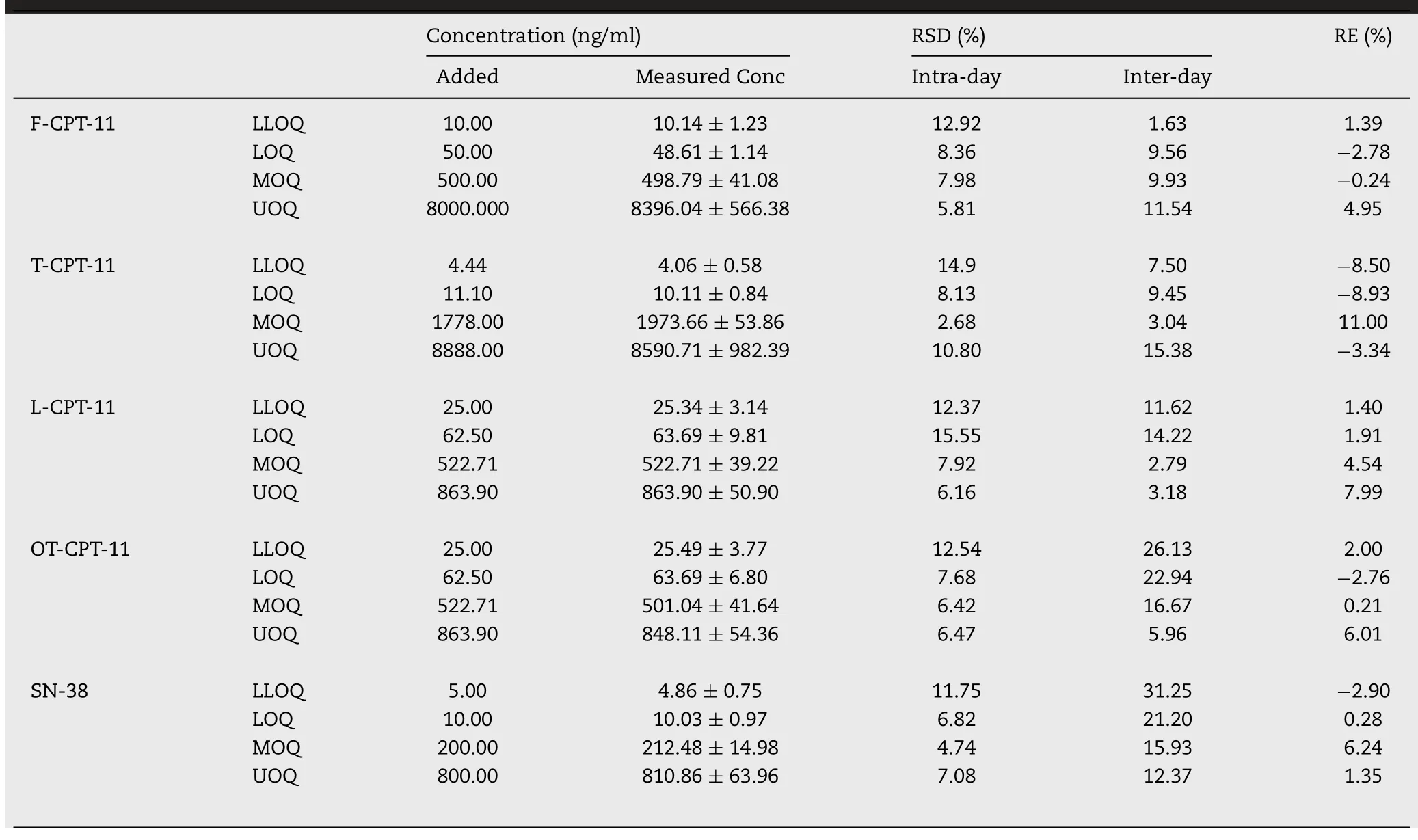
Table 1.-Precision and accuracy for assay of F-CPT-11,T-CPT-11,SN-38 in rat plasma and L-CPT-11,OT-CPT-11 in mouse at QC concentrations on three consecutive days(mean±SD,n=6).
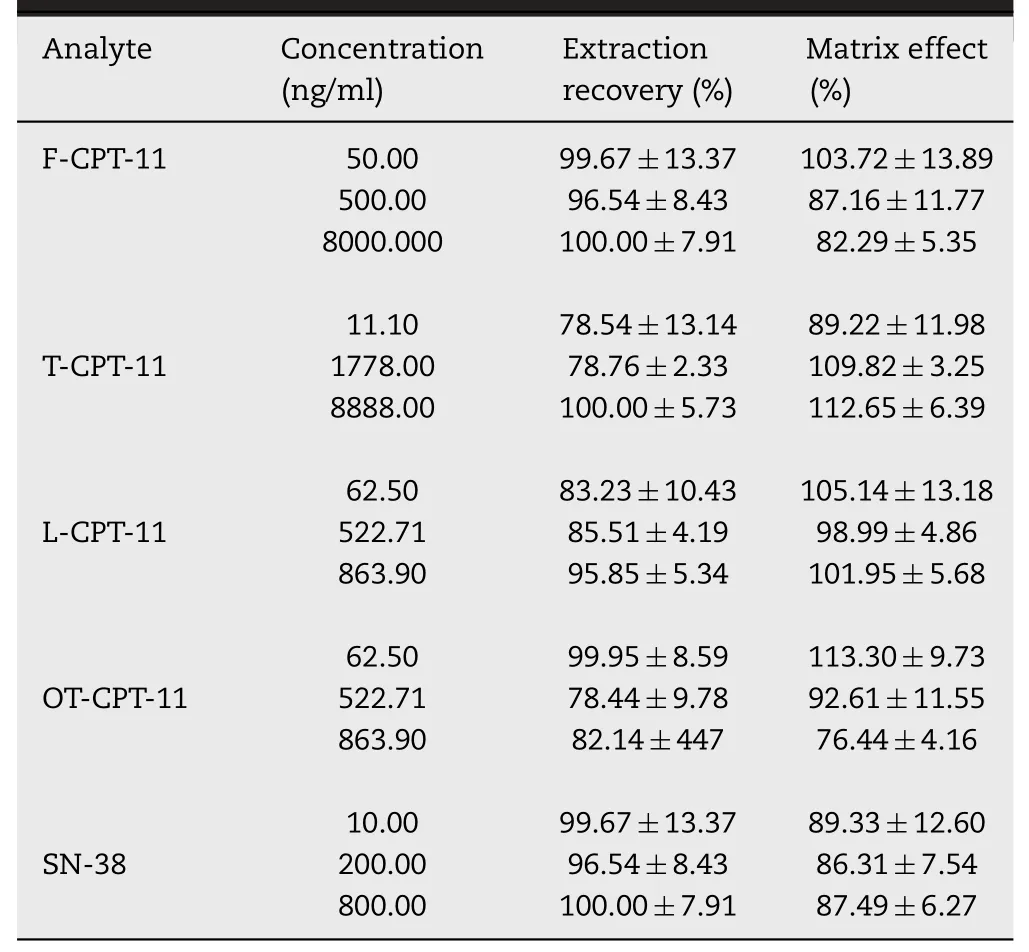
Table 2.-Matrix effect and extraction recovery of F-CPT-11,T-CPT-11,L-CPT-11,SN-38 in rat plasma and L-CPT-11,OT-CPT-11 in mouse at QC concentrations(mean±SD,n=18).
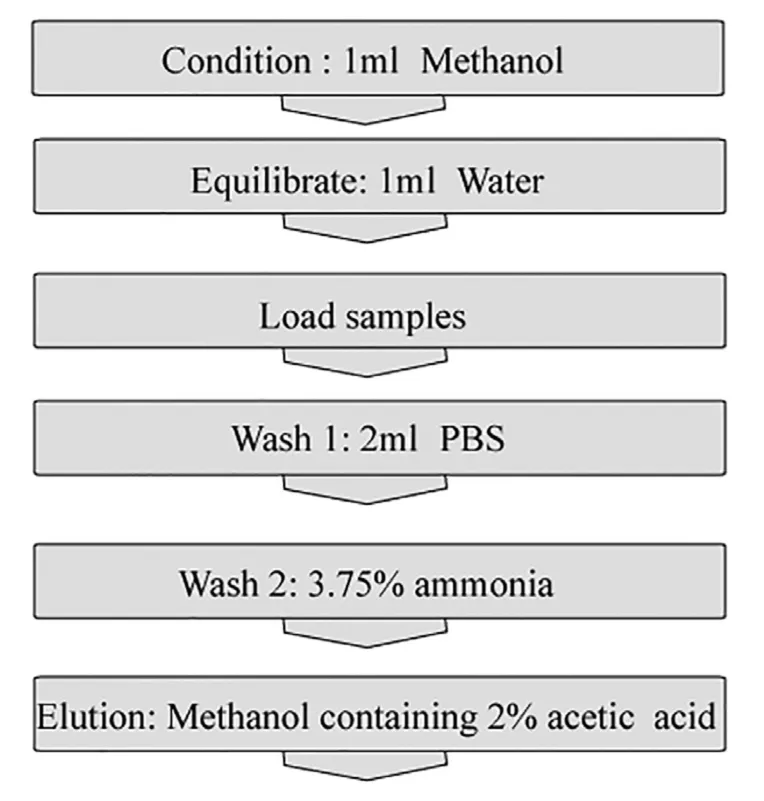
Fig.3.-Protocol for separating F-CPT-11 from plasma by SPE.

Table 3.-Method feasibility of QC samples for SPE process.
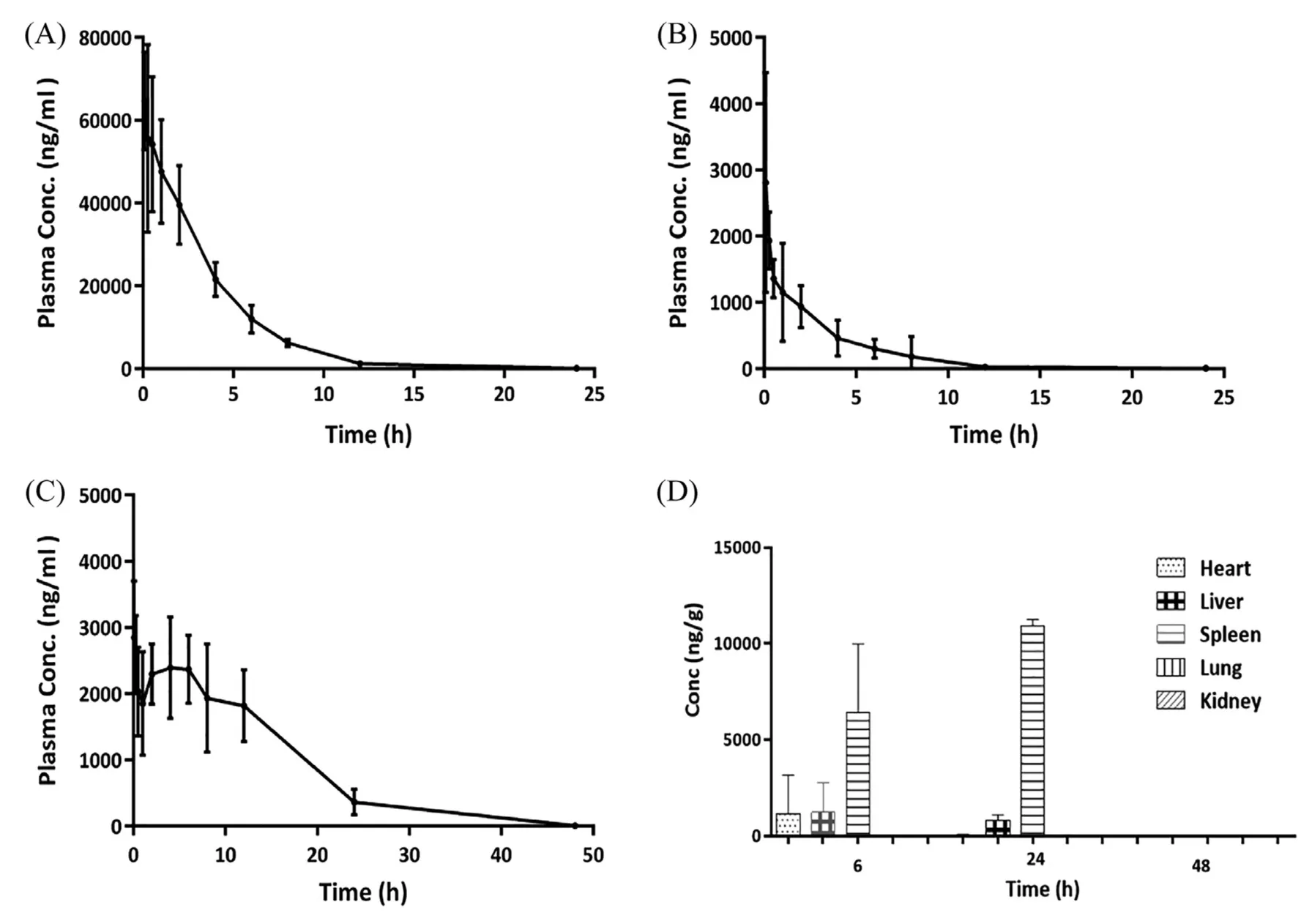
Fig.4.-Plasma concentration profiles of T-CPT-11(A),F-CPT-11(B),SN-38(C)after intravenous administration of 2.5 mg/kg CPT-11 liposome to rats and tissue distribution of CPT-11 after intravenous administration of 2.5 mg/kg CPT-11 liposome to mice(D).
3.4.Preparation and characterization of CPT-11 liposome
For CPT-11 liposome formulation,the entrapment efficiency was 98.23%.The mean diameter and zeta potential of CPT-11 liposomes measured by DLS were 131.4±0.14 nm and-18.5±8.5 mV,respectively.
3.5.Pharmacokinetic and bio-distribution study
The method described above was then used for pharmacokinetic and bio-distribution study after intravenous dose of 2.5 mg/kg CPT-11 liposome.The plasma concentrations-time curves of F-CPT-11,T-CPT-11 and SN-38 were presented in Fig.4.And main pharmacokinetic parameters of the analytes were shown in Table 4.After injection of CPT-11 liposomes,it was anticipated that most of the CPT-11 measured in rat plasma was encapsulated in the liposomal carrier to avoid system toxicity[11].In this study,theCmaxof F-CPT-11,TCPT-11 were 1979.37 ng/ml and 45 812.62 ng/ml respectively,while the latter is about 23 times higher than the former.At 30 min after intravenous administration of CPT-11 liposome,the plasma concentration of F-CPT-11 was 1359.5 ng/ml,and that of T-CPT-11 was 54 207.36 ng/ml.This result indicated that F-CPT-11 represented 2.5% of the total CPT-11 in the plasma at this time point.As the result of method fea-sibility above,we believe that leakage of liposome(below 5%)during the SPE process was acceptable,and SPE could be used to separate F-CPT-11 from liposome without changing the percentage of liposome artificially.The proportion of F-CPT-11 in plasma was also assessed by comparing the AUC values for both drug forms.The exposure amount(AUC0-48 h)of T-CPT-11 was found about 42 times higher than that of FCPT-11 plasma,which means liposomalization and PEG modification significantly increased the circulation time of CPT-11.Previous study have shown that enzyme in rats plasma can catalyze the conversion of CPT-11 to SN-38[24].AndMRT(0-48h)of SN-38 to that of F-CPT-11 ratio in this study was found to be 2.68 and long retention time of SN-38 could be explained by(1)entrapped CPT-11 in liposomes were stable in rat serum(2)CPT-11 was slowly released from liposomes,and then rapidly converted into SN-38 with catalysis of enzyme.

Table 4.-Pharmacokinetic parameters of F-CPT-11,T-CPT-11,L-CPT-11 and main metaboliteSN-38 in rat plasma after intravenous administration of 2.5 mg/kg CPT-11 liposome,(mean±SD,n=5 for T-CPT-11 and SN-38,n=3 for F-CPT-11).
CPT-11 levels in tissues determined at 6 h,24 h and 48 h were shown in Fig.4D.The highest concentration of CPT-11 were observed in spleen and followed by liver.And in heart,the concentration of CPT-11 fell off quickly during 6 h to 24 h after administration.The bio-distribution results indicated that CPT-11 liposome was more efficient in delivering CPT-11 to spleen and liver compared with other tissues.
4.Conclusion
In this study,a selective and sensitive UPLC-MS/MS method coupled with SPE technique was developed to quantify the F-CPT-11 and T-CPT-11 in rat plasma.And SPE method was feasible for separating F-CPT from liposome.The UPLC-MS/MS method was then successfully applied for pharmacokinetic and bio-distribution studies of CPT-11 liposome.The result showed that the validated method would be helpful for toxicity assessment of CPT-11 loaded-liposome.
Conflicts of interest
The authors declare no competing financial interest.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2018.08.003.
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Cancer nanotechnology:Enhancing tumor cell response to chemotherapy for hepatocellular carcinoma therapy
- The functions and applications of A7R in anti-angiogenic therapy,imaging and drug delivery systems
- Investigation of molecular aggregation mechanism of glipizide/cyclodextrin complexation by combined experimental and molecular modeling approaches
- Evaluation of the Mrp2-mediated flavonoid-drug interaction potential of quercetin in rats and in vitro models
- A novel oral prodrug-targeting transporter MCT 1:5-fluorouracil-dicarboxylate monoester conjugates
- Synthesis,characterization and in vivo evaluation of honokiol bisphosphate prodrugs protects against rats’brain ischemia-reperfusion injury