Evaluation of the Mrp2-mediated flavonoid-drug interaction potential of quercetin in rats and in vitro models
2020-01-06
aDivision of Biopharmaceutics,College of Pharmacy,Kyung Hee University,Seoul 02447,Republic of Korea
bDepartment of Life and Nanopharmaceutical Sciences,College of Pharmacy,Kyung Hee University,Seoul 02447,Republic of Korea
ABSTRACT Quercetin is a biologically active flavonoid that has been used as a popular health supplement.It is reported that quercetin may cause flavonoid-drug interaction mediated by P-glycoprotein,the most predominant efflux transporter.In this study,we comprehensively evaluated the potential of the pharmacokinetic interaction of quercetin mediated by multidrug resistance-associated protein 2(MRP2),another major efflux transporter.MRP2-transfected MDCKII cells and LS174T cells were used to evaluate the potential inhibition and induction of MRP2 by quercetin in vitro.To evaluate the induction effect of quercetin on Mrp2 in vivo,Mrp2 mRNA expression in rat liver,kidney,and small intestinal tissues was determined after the oral administration of quercetin(50,100,or 250 mg/kg)for seven days.Mrp2-mediated interaction potential was also evaluated by the pharmacokinetic study of phenolsulfonphthalein in rats after single or multiple doses of quercetin.Additionally,the effect of quercetin on absorption of docetaxel,a P-glycoprotein and CYP3A4 substrate,was also evaluated.Quercetin inhibited the function of MRP2 at 10 μM and induced the mRNA expression of MRP2 at 50 μM in vitro.Additionally,at 100 mg/kg,quercetin markedly increased Mrp2 expression in the small intestine of rats.However,there was no significant change in phenolsulfonphthalein pharmacokinetics due to single-(50,100,or 250 mg/kg)or multiple-dose(50,100,or 250 mg/kg for seven days)quercetin co-administration.By contrast,a significant interaction caused by quercetin(100 mg/kg)was observed in the absorption of docetaxel.The results suggested that although quercetin modulates the function and expression of MRP2 in vitro,it may have a low potential of Mrp2-mediated interaction and present negligible safety concerns related to the interaction.
Keywords:Quercetin P-glycoprotein Multidrug resistance-associated protein 2 Pharmacokinetics Flavonoid-drug interaction
1.Introduction
Quercetin is a flavonoid that exists abundantly in the glycosidated form in various fruits and vegetables,such as apples,onions,capers,and red wine.The daily dietary intake of quercetin is 5-40 mg;however,around 200-500 mg of quercetin can be consumed in a day in high-end consumers[1].Various bioactivities of quercetin have been supported byin vitro,preclinical,and epidemiological studies.Health supplements containing quercetin aglycone have been used at doses up to 1500 mg/d[2].Rutin(quercetin-3-O-rutinoside)is another commercially available source of quercetin[3].
Herb-drug or flavonoid-drug interactions can compromise the efficacy and safety of co-administered drugs.Drug metabolizing enzymes have mainly been investigated in this regard;however,drug transporters that are major determinants of plasma or tissue drug exposure have recently gained attention.In particular,there has been a growing interest in the interaction between herbs/flavonoids and efflux transporters,such as P-glycoprotein(P-gp)and multidrug resistance-associated proteins(MRPs),because they may directly affect the oral absorption and tumor distribution of anticancer agents[4].
Quercetin is reported to cause flavonoid-drug interactions by modulating the function and/or expression of metabolic enzymes.Quercetin induces the mRNA expression of CYP3A4 by about two-fold in human hepatocytes[5].In addition,the activity of CYP3A4,CYP2C8,and CYP2C9 decreases by quercetin treatment,whereas the activity of CYP1A1 is increased[6,7].Moreover,it clinically decreases and increases CYP1A2-and 2A6-mediated caffeine metabolism,respectively[8].
In a previous study,after the co-administration of quercetin(50 mg/kg)and digoxin in pigs,two-thirds of the pigs died,and the remaining one showed signs of acute poisoning to digoxin due to increased digoxin concentration as a result of P-gp inhibition by quercetin;it was considered as an example of fatal pharmacokinetic flavonoid-drug interactions[9].In addition,quercetin inhibited the efflux of taxol and irinotecan in Caco-2 cells[10,11].It increased the oral absorption rate of irinotecan and decreased the biliary excretion of its toxic metabolite in a preclinical study[10].These results suggest the potential use of quercetin in combination with anticancer agents to increase the efficacy and safety of chemotherapeutic regimens.
Quercetin is also reported to have an inducing effect on transporters.mRNA and protein expression of MRP1 was increased when MCF-7 cells were treated with 50 and 100 μM quercetin for 48 h,and the protein expression of UGT1A6 and MRP2 was increased in Caco-2 cells after treatment with quercetin for 72 h[12,13].
Mrp2 it has been considered a potential source of herbdrug interactions.Grape juice,Scutellariae Radix extract,Rhei Rhizoma extract,praeruptorin,and Inchinkoto have been reported to cause pharmacokinetic interactions that are associated with MRP2/Mrp2[14-18].Despite the possibility of the interaction of quercetin with Mrp2,follow-up studies were not conducted properly.In this study,we evaluated comprehensively the effect of quercetin on the inhibition and induction of Mrp2 usingin vitromodels and anin vivorat model.
2.Materials and methods
2.1.Materials
Quercetin dihydrate,phenolsulfonphthalein(PSP),probenecid,rifampicin,vincristine,Hank’s balanced salts,minimum essential medium(MEM)non-essential amino acid solution,poly-L-lysine hydrobromide,D-glucose,HEPES,and sodium bicarbonate were purchased from Sigma(St.Louis,MO).Docetaxel trihydrate was obtained from Shin Poong Pharmaceutical Co.Ltd.(Ansan,Republic of Korea).Zoletil®50(a mixture of 25 mg/ml of zolazepam and tiletamine each)was obtained from Virbac(Carros,France).All other chemicals and reagents were of analytical or HPLC grade as appropriate.
2.2.Uptake assay in MDCKII-WT and MDCKII-MRP2 cells
MDCKII-mock(MDCKII-WT)and MRP2-transfected MDCKII(MDCKII-MRP2)cell lines were kindly provided by Dr.Piet Borst(The Netherlands Cancer Institute)[19]and were grown in MEM supplemented with 1×MEM non-essential amino acid,10 mM HEPES,2 mM L-glutamine,100 units/ml penicillin,100 μg/ml streptomycin,and 10%FBS at 37°C in a humidified 5%CO2atmosphere[19].After two days of seeding at a density of 5×104cells/ml,cells were washed twice with 1×PBS and were incubated in a transport media consisting of 1×Hank’s balanced salts solution,10 mM HEPES,4 mM sodium bicarbonate,and 10 mM glucose.After 30 min,cells were incubated in 1 mM PSP following pretreatment with the test compound(1 mM probenecid in the transport media,and 5 and 10 μM quercetin in 0.5% DMSO)for 15 min.Cellular uptake was stopped after 90 min by washing the cells four times with ice-cold PBS.After lysis using 1 N NaOH,cell lysates were neutralized by adding 1 N HCl and subjected to HPLC analysis as previously reported[20].
2.3.mRNA quantification and efflux assay in LS174T cells
LS174T cell line was obtained from the Korean Cell Line Bank(Seoul,Republic of Korea)and cultured in RPMI1640 supplemented with 100 units/ml penicillin,100 μg/ml streptomycin,and 10% FBS at 37 °C in a humidified 5% CO2atmosphere.Cells were seeded at a density of 2×105cells/ml(for RNA extraction)and 1×105cells/ml(for efflux assay onto culture plates coated with poly-L-lysine)and were incubated in a culture medium containing rifampicin(10 μM),vincristine(5 and 10 nM),and quercetin(5,10,and 50 μM in 0.5% DMSO);the medium was replaced every 24 h.After 48 h,total RNA was isolated and the calcein efflux assay was conducted according to the manufacturer’s protocol(RNAiso,Takara,Japan)and a previous report,respectively[21].cDNA was synthesized using Takara PCRTMKit(AMV)ver.3.0,and mRNA expression was quantified using a LightCycler ver.1.5(Roche,Germany)and SYBR Premix Ex Taq(Takara,Japan).The mRNA levels were calculated by relative quantification with glyceraldehyde-3-phosphate dehydrogenase mRNA level as the endogenous control.The effect of quercetin on calcein efflux via MRP2 was evaluated based on the ratio of calcein level in the media to that in the cells.After drug treatment for 48 h as described above,calcein level in the media and in cells was quantified after incubation with 1 μM calcein-AM at 4 °C for 20 min followed by incubation in the transport media at 37°C for 1 h.
2.4.mRNA quantification in the liver,kidney,and small intestine of rats
Quercetin(50,100,and 250 mg/kg in 0.5% Na-carboxymethyl cellulose)or vehicle was orally administered for seven consecutive days to male Sprague-Dawley rats obtained from Samtako(Osan,Republic of Korea)and liver,kidney,and small intestinal tissues were excised on the 8 d.RNA extraction and real-time qPCR were carried out as described in the section of“2.3.mRNA quantification and efflux assay in LS174T cells”.mRNA levels were calculated by relative quantification with 18s RNA as the endogenous control.
2.5.Pharmacokinetic study of PSP in rats
Male Sprague-Dawley rats(220-300 g)obtained from Samtako(Osan,Republic of Korea)were housed at 25 °C and a 12-h light/dark cycle and had free access to food and water,except 12 h fasting before the experiment.For the single-dose administration study,quercetin(50,100,and 250 mg/kg)or vehicle was orally administered 1 h before PSP administration.For the multiple-dose administration study,rats were pretreated in the same manner as mentioned in the section of“2.4.mRNA quantification in the liver,kidney,and small intestine of rats”.Under anesthesia with 1 ml/kg of Zoletil®50(IP),femoral artery/vein,bile duct,and urinary bladder were cannulated with polyethylene tubes and PSP was intravenously injected at a dose of 0.8 mg/kg.Mannitol solution(4%,w/v)was infused at a rate of 2 ml/h via the femoral vein as an osmotic diuretic.Blood samples were collected from the femoral artery and centrifuged at 16 850×gfor 3 min.Bile and urine samples were collected in pre-weighed tubes and bile flow rate was measured gravimetrically assuming a density of 1.0.All samples were stored at-80 °C until analysis.PSP quantification was performed as previously reported using HPLC[20].The experimental protocol of the animal study was approved by the Committee on Care and Use of Laboratory Animals of Kyung Hee University.
2.6.Pharmacokinetic study of docetaxel in rats
After fasting for 12 h,male Sprague-Dawley rats were anesthetized with ether and surgical process was performed as described previously[22].After awakening from anesthesia,rats were orally administered quercetin alone or a mixture with docetaxel.The doses of quercetin and docetaxel were 100 mg/kg and 40 mg/kg,respectively.Blood samples were collected from carotid artery at the designated time for 10 h.The plasma concentration of docetaxel was quantified by LCMSMS analysis as previously reported[22].
2.7.Pharmacokinetic analysis
Pharmacokinetic parameters of PSP and docetaxel were determined using the model-independent method.The equations for area under the plasma concentration-time curve from time zero to infinity(AUCinf),clearance(CL),the volume of distribution at steady-state(Vdss),mean residence time(MRT),biliary clearance from plasma to bile(CLbile),and urinary clearance from plasma to urine(CLurine)were as follows:
AUCinf=AUClast+AUCterminal
AUCterminal=Cp,last/k
CL=Dose/AUCinf
Vdss=MRT×CL
MRT=AUMCinf/AUCinf
CLbile=Xbile,120min/AUClast
CLurine=Xurine,120min/AUClast
where AUClast,Cp,last,k,AUMCinf,Xbile,120min,andXurine,120min,represent the AUC from time zero to the last sampling time point calculated using the linear trapezoidal method,the plasma concentration of PSP or docetaxel at the last sampling time point,the slope of the terminal phase of the plasma concentration-time curve,the area under the first-moment curve of the plasma concentration-time curve from time zero to infinity,the excreted amount of PSP in the bile for 120 min,and the excreted amount of PSP in the urine for 120 min,respectively.
2.8.Statistical analysis
Statistical significance was analyzed using SPSS 15.0 program.Unpairedt-test was applied to analyze data obtained fromin vitroexperiments using MDCKII and LS174T cells andin vivopharmacokinetic study of docetaxel.Differences in the mRNA expression and pharmacokinetic parameters of PSP between control and quercetin-treated rats were verified using one-way ANOVA followed by LSD and Dunnett’spost hoctest,respectively.A value ofP<0.05 was considered statistically significant.
3.Results and discussion
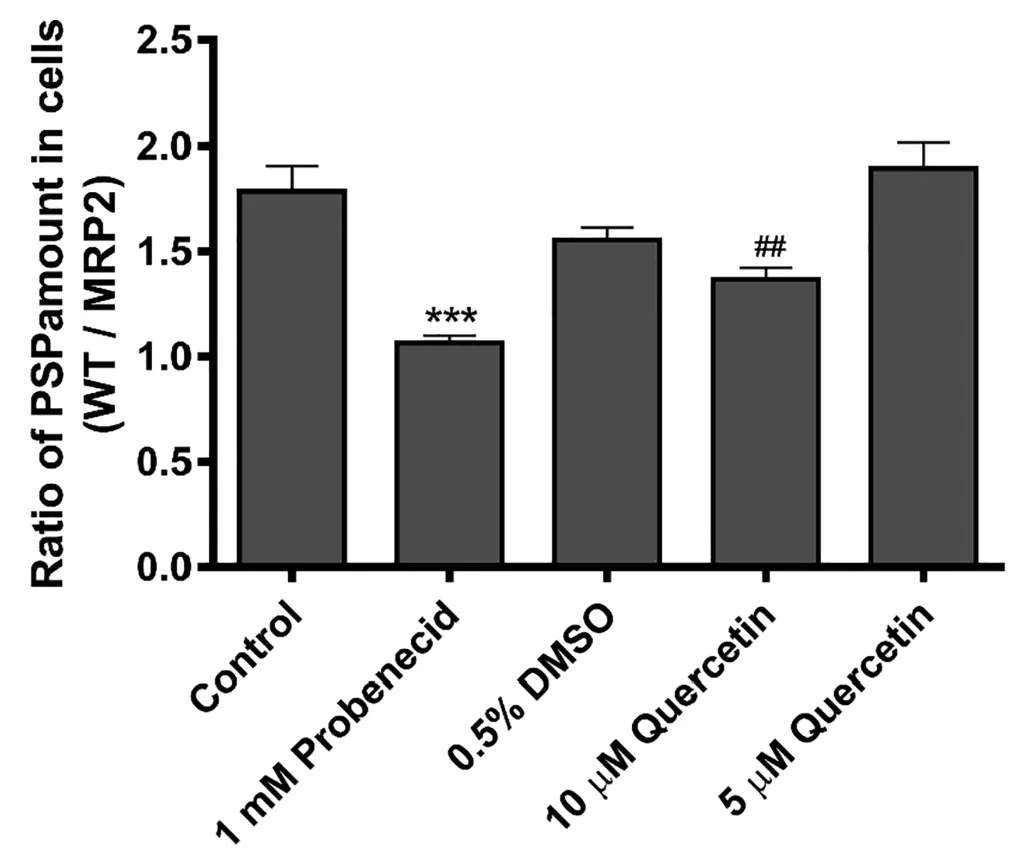
Fig.1-Effects of quercetin on the membrane transport of phenolsulfonphthalein via multidrug resistance-associated protein 2(MRP2).Cells were pretreated with the transport media(control,n=3),0.5%DMSO(n=3),1 mM probenecid(n=3),10 μM quercetin(n=6),or 5 μM quercetin(n=6)for 15 min followed by incubation in 1 mM phenolsulfonphthalein for 90 min.The transport media and 0.5%DMSO were used as vehicle controls for probenecid and quercetin treatment groups,respectively.Data are expressed as the mean±SD for the ratio of phenolsulfonphthalein amount in MDCKII-WT to that in MDCKII-MRP2;∗∗∗P<0.001 vs.control(transport media only),##P<0.01 vs.control(0.5%DMSO).
The uptake of PSP,a typical MRP2 substrate,into MDCKII-MRP2 was low compared to that into MDCKII-WT.In addition,the ratio of the intracellular concentration of PSP in MDCKII-WT to that in MDCKII-MRP2 cells was significantly decreased upon exposure to the typical MRP2 inhibitor,probenecid.These results confirmed that PSP is a substrate of MRP2[23].In this MRP2 assay system,quercetin showed a significant inhibitory effect on MRP2 at a concentration of 10 μM,which was lower concentration compared to the previous results(Fig.1)[24].
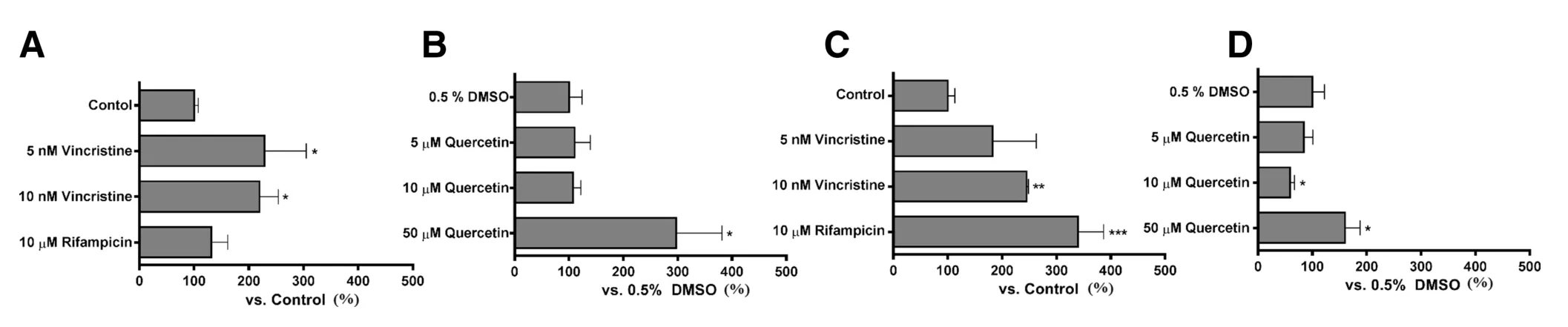
Fig.2-(A)and(B)mRNA expression of multidrug resistance-associated protein 2 and(C)and(D)MDR1 in LS174T cells after treatment with(A)and(C)known inducers and(B)and(D)quercetin for 48 h.Rifampicin and vincristine were used as positive controls and 0.5%DMSO was used as the vehicle for quercetin.Data are expressed as the mean±SD for percentage compared to the vehicle-treated group;∗P<0.05,∗∗P<0.01,∗∗∗P<0.001 vs.control or 0.5%DMSO(n=3).
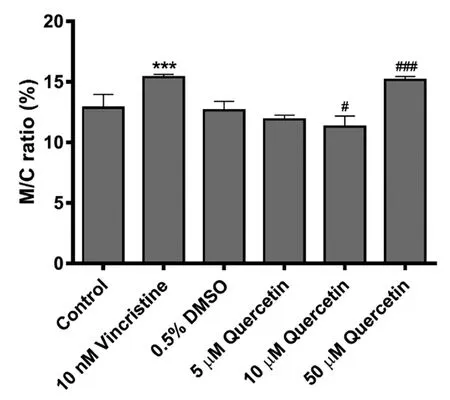
Fig.3-Media-to-cell(M/C)ratio of calcein amount in LS174T cells.Data are expressed as the mean±SD;∗∗∗P<0.001 vs.control;#P<0.05,###P<0.001 vs.0.5%DMSO(n=6).
LS174T,human colon carcinoma cell line,has been used to evaluate the induction of drug transporters by xenobiotics[25].In this study,we investigated the effect of quercetin on the expression of MRP2 and P-gp(MDR1).Vincristine(10 nM)and rifampicin(10 μM)were used as known inducers of MRP2 and MDR1,respectively(Fig.2A and C)[25].Quercetin(50 μM)increased MRP2 mRNA expression by 3.0-fold compared to control,and this increase was larger than that caused by vincristine(Fig.2B).Quercetin(50 μM)also increased MDR1 mRNA expression(Fig.2D).
The calcein-AM assay was performed to investigate whether the increase in MRP2 mRNA expression resulted in a change in transport activity.Calcein-AM is an acetoxymethylester derivative of calcein,which itself does not fluoresce,but is converted to fluorescent calcein,an MRP2 substrate,by intracellular esterase after cellular uptake[26].As with the positive control,vincristine,quercetin(50 μM)increased the ratio of excreted calcein to extracellular calcein(media-to-cell ratio,a marker of MRP2 activity)(Fig.3).Statistically different media-to-cell ratio was observed in quercetin(10 μM)treatment group,but absolute difference was small(10.7% of control).These results on mRNA expression and membrane transport suggest that long-term exposure to quercetin modulates the gene expression and function of MRP2 at the cellular level.Although the concentrations usedin vitromodels could be considered as relatively high considering thein vivoplasma concentration of quercetin,luminal quercetin concentration will be much higher than its plasma concentration.The concentration of xenobiotics in enterocytes could depend on the luminal concentration,not on the systemic concentration.This could be the reason for the significant induction in the intestinein vivo,which is described below.
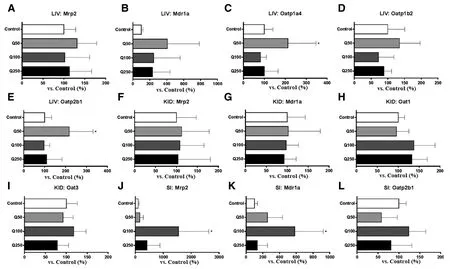
Fig.4-(LIV;A-E)mRNA expression of drug transporters in the liver,(KID;F-I)kidney,and(SI;J-L)small intestine.Rats were orally administered 50(Q50),100(Q100),or 250 mg/kg(Q250)of quercetin in 0.5%Na-carboxymethyl cellulose for seven consecutive days and mRNA was isolated on the 8 d.Data are expressed as the mean±SD for percentage compared to the vehicle-treated group(control);∗P<0.05 vs.control(n=3-6).
Multiple-dose quercetin administration(50,100,and 250 mg/kg)for seven days did not alter the mRNA expression of rat Mrp2 and rat Mdr1a,the ortholog of human MDR1,in the liver and kidney(Fig.4A,4B,4F,and 4G).However,it increased the intestinal mRNA expression of rat Mrp2 and rat Mdr1a(Fig.4J and 4K).The most dramatic change in mRNA expression was observed at the dose of 100 mg/kg;Mrp2 and Mdr1a expression was increased by 15.4-and 5.8-fold,respectively.It would be interesting to investigate whether intestinal drug absorption or secretion via Mrp2 or P-gp is affected by multiple-dose quercetin administration.Because quercetin dramatically altered Mrp2 level,intestinal drug secretion via Mrp2 was evaluated indirectly by evaluating PSP pharmacokinetics after multiple-dose quercetin administration in rats.
Quercetin can be taken up to 2000 mg/d as a health supplement.Considering body surface area,the human equivalent dose for a rat dose of 100 mg/kg corresponds to 1129 mg/70 kg[27];therefore,it is difficult to say that the 100 mg/kg is extremely high dose.
When exposed to quercetin for seven days,the mRNA expression of Mrp2 and Mdr1a in tissues showed different patterns depending on the dose and tissue.There was no apparent change in their expression in the liver and kidney.This could be because the intestines were exposed to high concentrations of quercetin in the intestinal lumen,whereas the direct exposure of liver or kidney to quercetin was limited by first pass effect.
The induction effect on transporters was most pronounced at the middle dose of 100 mg/kg.A similar biphasic biological effect of quercetin has been reported previously,in which the effect of quercetin on the ATP hydrolysis of MRP1 and MRP4 was bilateral.Quercetin promoted ATPase activity at a low concentration and inhibited the activity at a high concentration[28].At a low concentration,quercetin and kaempferol inhibited the uptake of vincristine by P-gp,but an increased uptake of vincristine was observed at a high concentration[29].

Fig.5-(A)Plasma concentration-time profile,(B)cumulative biliary excretion after 2 h,and(C)urinary excretion after 2 h of phenolsulfonphthalein administration in rats.Rats were intravenously injected 0.8 mg/kg of phenolsulfonphthalein after 1 h of the oral administration of 50,100,and 250 mg/kg of quercetin in 0.5%Na-carboxymethyl cellulose.●,◆,▼,and ▲ indicate vehicle-(control,n=7),50(Q50,n=6),100(Q100,n=6),and 250 mg/kg(Q250,n=5)quercetin-treated groups,respectively.Data are expressed as the mean±SD;no statistical differences were observed.
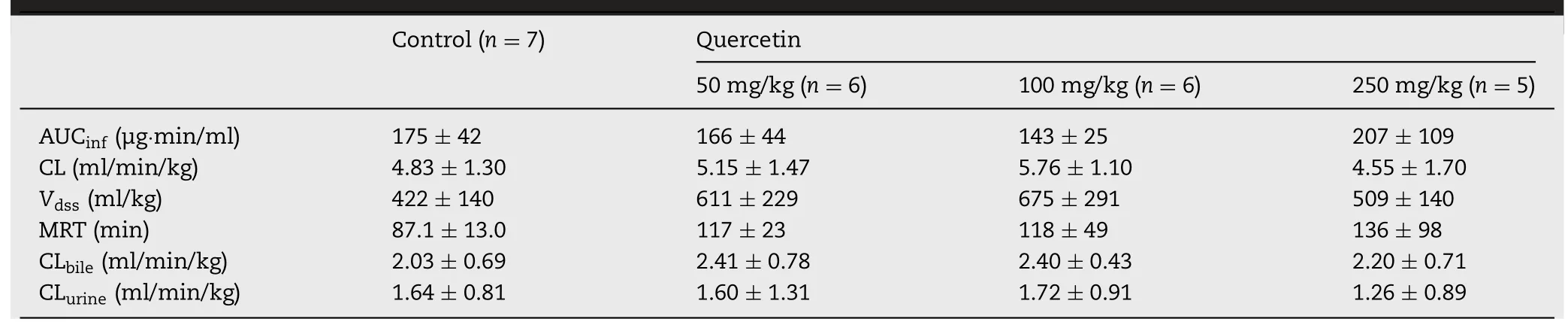
Table 1-Pharmacokinetic parameters of phenolsulfonphthalein in rats after the single oral administration of quercetin.Data are expressed as the mean±SD;no statistical differences were observed.
The organic anion transporting polypeptide(OATP)family are mainly distributed in the liver and contribute to the hepatic uptake of various types of organic anions[30].Similarly,the organic anion transporter(OAT)family of transporters present in the kidney.Although the fold increase in the expression of Oatp1a4 and Oatp2b1 was lower than that in the expression of Mrp2 in the intestinal tissue,the expression of Oatp1a4 and Oatp2b1 was increased by 2.1-and 2.2-fold,respectively,in the liver after exposure to 50 mg/kg quercetin(Fig.4C and 4E).This finding suggests that continuous quercetin administration may increase the hepatic uptake of OATP substrates.However,mRNA levels at doses higher than 50 mg/kg were similar to control,and Oatp1b2 mRNA level was not changed in all three dosing groups(Fig.4D).
The expression of Oat1 and Oat3 in the kidney(Fig.4H and 4I)and Oatp2b1 in the intestine(Fig.4L)was not affected by quercetin.
To support results of mRNA induction,the protein expression of these transporters was not checked in this study.We expected that anin vivopharmacokinetic study after multipledose quercetin administration in rats can be used to detect functional changes in these transporters because PSP is a cosubstrate of MRP2/Mrp2,Oatp1B1,Oat1,and Oat3[31,32].
The inhibitory effect of quercetin on Mrp2in vitrowas also evaluated by anin vivopharmacokinetic study of PSP in rats.Interestingly,the plasma concentration profile,biliary excretion,and urinary excretion of PSP when co-administered with quercetin(50,100,and 250 mg/kg)were similar to those in the absence of quercetin(Fig.5),and pharmacokinetic parameters of PSP were not affected by quercetin(Table 1).Because Mrp2 activity is known to affect the biliary,urinary,and intestinal secretion of PSP[20,33],these results suggest that the effect of quercetin absorbed after single oral administration on the excretion of Mrp2-mediated drugs would be small due to significant first pass effect.More than 90%of orally administered quercetin is metabolized during absorption,and only about 5.3% is observed in the systemic circulation[34].Although we did not measure the plasma concentration of quercetin,Cmaxafter the oral dosing of quercetin at 250 mg/kg is estimated at 1.8 μg/ml(6.0 μM)from a previous pharmacokinetic study[35].Because quercetin metabolites also inhibited MRP2,the interpretation of results of this study became more difficult.For example,3′-O-methylquercetin and 7-O-glucuronosyl quercetin,phase II metabolites of quercetin,significantly inhibit calcein uptake via MRP2[36],whereas total metabolites of quercetin after incubation with rat S9 fraction does not inhibit P-gp function[37].
Generally,flavonoids predominantly exist in the form of glucuronidated and sulfated(mixed or multiple)conjugates in the blood.Glucurono-sulfo-conjugated quercetin and glucurono-sulfo-conjugated isorhamnetin (3′-Omethylquercetin)are the dominant forms(more than 90%)found in rats[38,39].
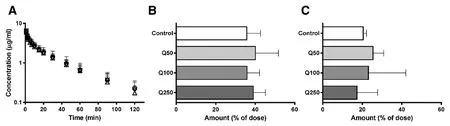
Fig.6-(A)Plasma concentration-time profile,(B)cumulative biliary excretion after 2 h,and(C)urinary excretion after 2 h of phenolsulfonphthalein administration in rats.Rats were intravenously injected 0.8 mg/kg of phenolsulfonphthalein after the oral administration of 50,100,and 250 mg/kg of quercetin in 0.5%Na-carboxymethyl cellulose for seven consecutive days.Pharmacokinetic experiments were performed on the 8th day.○,◇,▽,and △ indicate vehicle-(control,n=4),50(Q50,n=3),100(Q100,n=3),and 250 mg/kg(Q250,n=3)quercetin-treated groups,respectively.Data are expressed as mean±SD;no statistical differences were observed.
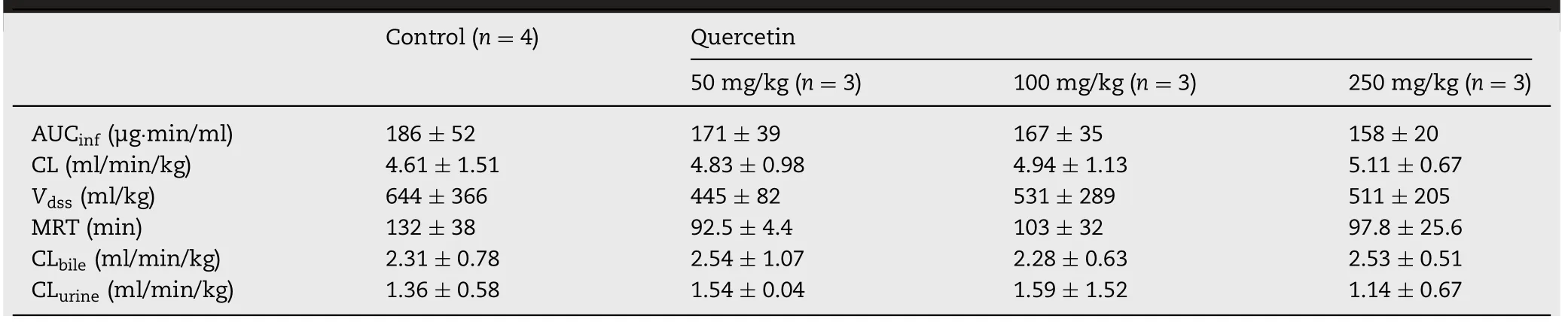
Table 2-Pharmacokinetic parameters of phenolsulfonphthalein in rats after the multiple oral administration of quercetin for seven days.Data are expressed as the mean±SD;no statistical differences were observed.
The reported plasma concentration of quercetin in humans after the administration of quercetin,rutin,or quercetin-containing diet varies substantially among studies.Because quercetin is extensively metabolized to glucuronide and sulfate conjugates,quercetin-3-glucuronide,3′-methylquercetin-3-glucuronide,and quercetin-3′-sulfate are majorly present in human plasma[40].TheCmaxof quercetin and its conjugates was 15.4 ng/ml(0.0509 μM)and 448 ng/ml(1.48 μM),respectively,after multiple-dose oral quercetin administration(500 mg t.i.d for seven days)[41].In addition,the plasma concentration of total quercetin(quercetin plus conjugated forms)after multiple-dose quercetin administration reached up 418 and 605 μg/l for 500 and 1000 mg/kg doses,respectively[42].Thus,it is important to identify the effect of quercetin metabolites in humans on MRP2 to relate this non-clinical data with the clinical situation.In addition,the high protein binding of quercetin should be considered[43]because it may decrease the free concentration of quercetin at the binding site of transporters.
To investigate the effect of quercetin on the hypothetical induction of transporters,quercetin(50,100,and 250 mg/kg)was administered as multiple doses for seven days,and changes in PSP pharmacokinetics were observed.The plasma concentration-time profile,biliary excretion,and urinary excretion of PSP when co-administered with quercetin were similar to those in the absence of quercetin(Fig.6),and pharmacokinetic parameters of PSP were not affected(Table 2).The expression of Mrp2 in the liver and Oat1 and Oat3 in the kidneys did not change after the administration of quercetin.Consequently,thein vivopharmacokinetics of PSP were not affected by quercetin.The two-fold increase in Oatp mRNA expression,which was observed in the 50 mg/kg quercetin group,did not affect PSP pharmacokinetics.Additionally,the plasma concentration-time profile of PSP did not change by multipledose quercetin administration,despite Mrp2 being involved in the intestinal secretion of PSP.This may be explained by inference that Oatps and Mrp2 were not induced at protein level.Otherwise,fold differences of induction were not great,and fail to affect plasma concentration profile of PSP.
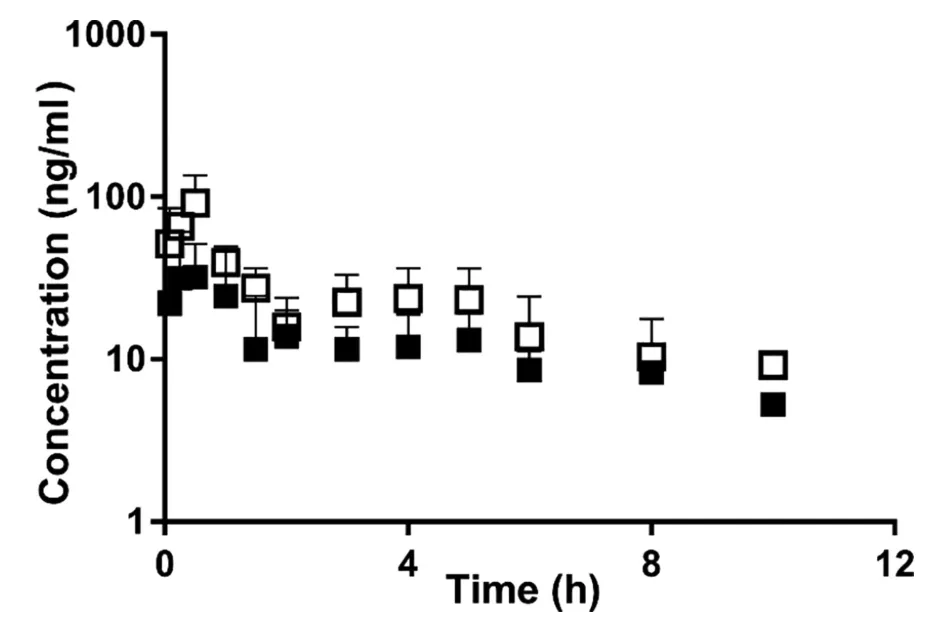
Fig.7-Plasma concentration-time profiles of docetaxel in rats.Rats were orally administered 40 mg/kg of docetaxel with or without 100 mg/kg of quercetin.■ and □ indicate control(n=4)and 100 mg/kg quercetin-treated group(n=3),respectively.Data are expressed as mean±SD.

Table 3-Pharmacokinetic parameters of docetaxel in rats after oral administration at the dose of 40 mg/kg of docetaxel with or without 100 mg/kg of quercetin.Data are expressed as the mean±SD;∗P<0.05 vs.control.
Meanwhile,plasma concentrations of docetaxel,a substrate for P-gp and CYP3A4[44],increased significantly by concomitant oral administration of quercetin,whereas quercetin administered orally did not affect the pharmacokinetics of PSP(Fig.7).AUCinfandCmaxof docetaxel in the quercetin-treated group were 1.8-and 2.5-fold higher than those in the control group,respectively(Table 3).The intestinal concentration of quercetin in this study was assumed to be about 18.1 mM,considering intestinal volume.Because it could be higher than IC50values of quercetin for P-gp and CYP3A4,both P-gp and Cyp inhibition could contribute to the increased the absorption of docetaxel[45-47].These results showed again that at least quercetin could affect pharmacokinetics of drugs when quercetin encounters victim drugs in the intestinal lumen.
Dosing formulation is another important factor influencing the plasma concentration of quercetin after its absorption[48].When quercetin-containing diet was fed to spontaneously hypertensive rats for 5 or 11 weeks(approximately equivalent to 20-40 mg/kg/d of quercetin),there was no delay in the development of hypertension compared with the control group.Whereas,rats administered quercetin orally(10 mg/kg)once a day for four days had lower blood pressures than control rats,suggesting that the effect of quercetin may depend on the dosage form[49].However,although we also evaluated PSP pharmacokinetics after an intravenous infusion of quercetin(147 mg/h/kg;maximum soluble dose)to exclude the absorption problem,there was no significant change in the Mrp2-mediated pharmacokinetics of PSP(data not shown).In this experiment,the total plasma concentration of quercetin at the steady-state was 23.6 μg/ml(equivalent to 78.1 μM).Because quercetin was reported to bind to plasma protein extensively,about 99%,its free plasma concentration was predicted to be much lower than the IC50value against MRP2 which was known to be higher than 50 μM[43].This might be the reason of inconsistent results betweenin vitroinhibition study andin vivostudy.In conclusion,considering the dose limitation due to low solubility,high protein binding,and the high IC50value of quercetin,a pharmacokinetic interaction between quercetin and MRP2 substrates should be negligible irrespective of the route of administration of quercetin.
4.Conclusion
In this study,we investigated the interaction between quercetin and Mrp2 throughin vitroexperiments andin vivopharmacokinetic studies.In conclusion,quercetin significantly interacts with Mrp2 at the cellular level.However,this result was not associated within vivoPSP pharmacokinetics.Multiple-dose quercetin administration resulted in an increase in the mRNA expression of transporters,including Mrp2 in the small intestine and Oatp in the liver.However,no changes in PSP pharmacokineticsin vivowere observed probably because quercetin existed mainly in its conjugated forms in the body.These results suggest that quercetin may be relatively safe in terms of Mrp2-mediated interaction.Another important lesson of this study is that bioavailability should be considered when we discuss flavonoid-drug interaction usingin vitroresults.Thus,the effects of metabolites present in the body should also be tested usingin vitroexperiments.
Conflicts of interest
The authors report no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science,ICT and Future Planning(NRF-2015R1A2A2A01002673).
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Cancer nanotechnology:Enhancing tumor cell response to chemotherapy for hepatocellular carcinoma therapy
- The functions and applications of A7R in anti-angiogenic therapy,imaging and drug delivery systems
- Investigation of molecular aggregation mechanism of glipizide/cyclodextrin complexation by combined experimental and molecular modeling approaches
- A novel oral prodrug-targeting transporter MCT 1:5-fluorouracil-dicarboxylate monoester conjugates
- Synthesis,characterization and in vivo evaluation of honokiol bisphosphate prodrugs protects against rats’brain ischemia-reperfusion injury
- Improved dissolution and oral absorption by co-grinding active drug probucol and ternary stabilizers mixtures with planetary beads-milling method