A novel oral prodrug-targeting transporter MCT 1:5-fluorouracil-dicarboxylate monoester conjugates
2020-01-06JinSun
,Jin Sun,
aWuya College of Innovation,Shenyang Pharmaceutical University,Shenyang 110016,China
bSchool of Pharmacy,Guangxi University of Chinese Medicine,Nangning 530200,China
cSchool of Pharmacy,Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016,China
dSchool of Life Science and Biopharmaceutics,Shenyang Pharmaceutical University,Shenyang 110016,China
ABSTRACT Monocarboxylate transporter 1(MCT1)is responsible for oral absorption of short-chain monocarboxylic acids from small intestine,hence,it’s likely to serve as an ideal design target for the development of oral prodrugs.However,potential application of MCT1 to facilitate the oral delivery is still unclear.Irregular oral absorption,poor permeability and bioavailability greatly limit the oral delivery efficiency of 5-fluorouracil(5-FU).Herein,we design three 5-FU-fatty acid conjugates targeting intestinal MCT1 with different lipophilic linkages.Interestingly,due to high MCT1 affinity and good gastrointestinal stability,5-FUoctanedioic acid monoester prodrug exhibited significant improvement in membrane permeability(13.1-fold)and oral bioavailability(4.1-fold)compared to 5-FU.More surprisingly,stability experiment in intestinal homogenates showed that 5-FU prodrugs could be properly activated to release 5-FU within intestinal cells,which provides an ideal foundation for the improvement of oral bioavailability.In summary,good gastrointestinal stability,high membrane permeability and appropriate intestinal cell bioactivation are the important factors for high-efficiency 5-FU oral prodrugs,and such work provides a good platform for the development of novel oral prodrugs targeting intestinal transporters.
Keywords:5-Fluorouracil Prodrugs Monocarboxylate transporter 1 Permeability Oral bioavailability
1.Introduction
5-Fluorouracil(5-FU)is widely used in the treatment of various solid tumors,such as colorectal cancer and breast cancer[1-3],and it is administrated by i.v.in clinic[4],which causes poor patient compliance and serious side effects[5,6].To address these issues,oral administration is more preferable.However,low membrane permeability across epithelium and rapid gastrointestinal(GI)degradation severely limit the development of oral 5-FU products[7,8].
In recent years,intestinal transporter-targeting prodrug strategy has attracted great attention for oral delivery due to increase in the intestinal permeability and GI stability and some prodrugs have entered the clinical trials[9-11].Early studies found that cytarabine 5′-valine ester could improve the oral bioavailability of cytarabine by targeting intestinal oligopeptide transporter 1(Pept1)[12],but its GI stability was poor.We also synthesized a novel oral prodrug by conjugating L-carnitine to the N4of gemcitabine to target intestinal organic cation transporter 2(OCTN2),with oral bioavailability approximately 4.9-fold higher than that of gemcitabine[13].More importantly,its GI stability was increased significantly compared to PEPT1-targeting prodrugs.However,the low activation efficiency of prodrugs in blood circulation system decreases an efficacy.Accordingly,concurrently optimizing permeability,stability and prodrug activation are very important for rational prodrug design.
Monocarboxylic acid transporter 1(MCT1)is critical in transporting monocarboxylic acid across the cell membrane[14,15],and is widely distributed throughout the intestines[16,17].Previous studies demonstrated that non-endogenous monocarboxylate andβ-lactam antibiotics could be absorbed through the intestine by MCT1[18].Tomoo Itoh et al.synthesized a hydrophobic conjugate of carbenicillin in order to improve the permeability by MCT1.The intestinal permeability of the hydrophobic conjugate of carbenicillin was 70-fold higher than carbenicillin and the improvement in lipid solubility of the prodrug was increased significantly,resulting in the lager contribution by passive diffusion over transportermediated active transport.Thus,the role of MCT1 as an oral prodrug designing target is still unclarified.
Herein,we conjugated di-carboxylic acids to 5-FU to synthesize MCT1-targeting prodrugs.Different linkers of fatty acids(hexanedioic acid,octanedioic acid and hexanoic acid)were introduced at N1position of 5-FU in order to achieve good membrane permeability and high oral bioavailability.The designed MCT1-targeting oral prodrugs were capable of coordinating different properties among good gastrointestinal stability,high permeability and moderate bioactivation,in order to meet different requirements onin vivoeach step of oral intestinal-transporter prodrugs.
2.Materials and methods
2.1.Materials
5-Fluorouracil(5-FU;99%)and 2-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate(HATU;99%)were purchased from Shanghai Macklin Biochemical Company(Shanghai,China).Adipic acid(98%),suberic acid(98%),n-hexanoic acid(98%)and quercetin(98%)were obtained from Shanghai Darui Chemical Co.,Ltd(Shanghai,China).Hexanedioic acid mono-(5-fluoro-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)ester(HDA-5-FU)and octanedioic acid mono-(5-fluoro-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)ester(OA-5-FU)and hexanoic acid-5-fluorouracil-1-methyl ester(HA-5-FU)were synthesized at Shenyang Pharmaceutical University.Caco-2 cells were obtained from ATCC(American Type Culture Collection).MEM/EBSS(minimum Eagle’s medium/Earle’s balanced salts solution)medium was obtained from Shenyang Xingboermei Trading Co.,Ltd(Shenyang,China).
2.2.Synthesis of prodrugs
The synthetic route of HDA-5-FU consisted of two steps.In the first step,5-fluorouracil(2.11 g,16.2 mmol)and 37%formaldehyde solution(8.04 ml,106.95 mmol)were added to a 50 ml eggplant-shaped flask,and stirred in 55°C oil bath to dissolve 5-fluorouracil.Stirring was then continued for 4 h.After completion of the reaction,a colorless transparent liquid was obtained,and excess water was distilled off under reduced pressure to give a colorless viscous liquid.In the second step,adipic acid(1.66 g,11.34 mmol),HATU(5.6 g,14.7 mmol)and TEA(triethylamine)(2.29 g,22.68 mmol)were placed in a 50 ml eggplant-shaped flask which containing 15 ml of dry acetonitrile and stirred in ice bath for 1 h.After 1 h,the product of the first step was dissolved in dry acetonitrile and added to the reaction solution,and then the reaction flask was reacted at room temperature for 5 h.After the reaction was completed,the reaction solution was distilled off under reduced pressure,and then liquid-liquid extraction was carried out with ethyl acetate.The organic layer was dehydrated by adding anhydrous sodium sulfate.Finally,the obtained crude product was purified by preparative HPLC.Chromatographic method was as followed:flow rate,5 ml/min;injection volume,1.5 ml,mobile phase consisted of a mixture of acetonitrile and water(35:65,v/v).After distillation under reduced pressure,a white powder was obtained.
For OA-5-FU,the first step of synthesis was the same as HDA-5-FU.In the second step,suberic acid(1.98 g,11.34 mmol),HATU(5.6 g,14.7 mmol)and TEA(2.29 g,22.68 mmol)were placed in a 50 ml eggplant-shaped flask which containing 15 ml of dry acetonitrile and stirred in ice bath for 1 h.The chromatographic methods were as follows:flow rate,5 ml/min;injection volume,1.5 ml,mobile phase consisted of acetonitrile and water(48:52,v/v).
For HA-5-FU,hexanoic acid(1.32 g,11.34 mmol),HATU(5.6 g,14.7 mmol)and TEA(2.29 g,22.68 mmol)were placed in a 50 ml eggplant-shaped flask which containing 15 ml of dry acetonitrile and stirred in ice bath for 1 h.The chromatographic methods were as follows:flow rate,5 ml/min;injection volume,1.5 ml,mobile phase consisted of acetonitrile and water(50:50,v/v).
HDA-5-FU:Purity:97%.MS(ESI):m/z287.3[M-H]-.1H NMR(600 MHz,DMSO)δ11.99(d,J=4.6 Hz,1H),8.13(d,J=6.5 Hz,1H),5.56(s,2H),2.36(t,J=7.0 Hz,2H),2.21(t,J=6.9 Hz,2H),1.57-1.45(m,4H).
OA-5-FU:Purity:97%.MS(ESI):m/z315.4[M-H]-.1H NMR(600 MHz,DMSO)δ12.03-11.94(m,2H),8.14(d,J=6.5 Hz,1H),5.56(s,2H),2.33(t,J=7.4 Hz,2H),2.18(t,J=7.4 Hz,2H),1.60-1.38(m,4H),1.31-1.20(m,4H).
HA-5-FU:Purity:96%.MS(ESI):m/z257[M-H]-.1H NMR(600 MHz,DMSO)δ11.99(s,1H),8.14(d,J=5.3 Hz,1H),5.57(s,2H),2.33(t,J=6.9 Hz,2H),1.58-1.45(m,2H),1.26(m,J=12.6 Hz,4H),0.85(t,J=6.1 Hz,3H).
2.3.Chemical and enzymatic stability evaluation
The chemical stability of 5-FU and prodrugs were carried out in different pH hydrochloride buffers and phosphate buffers(PBS)[19].Drugs were dissolved in different pH(1.2,6.8,7.4)to obtain prodrug solutions with concentration of 80 μg/ml.The drug-containing solutions were incubated in a shaker at 37°C,sampling at 0,0.083,0.25,0.5,1,2,4,8,12,24,48 h,centrifuged at 13 000 rpm for 10 min in a high-speed centrifuge.Detection was carried out by HPLC.
Enzyme stability was verified by rat(Sprague-Dawley)plasma and tissue homogenates.The rat plasma was placed in a-80°C refrigerator for use.Plasma was obtained from orbital blood samples.Liver and intestine of rat were washed with physiological saline,and tissues were ground to homogenates in an ice bath at a weight ratio of tissues to physiological saline of 1:3,and the supernatant was taken after low temperature centrifugation.Drugs were dissolved in rat plasma and tissue homogenates,obtaining a homogenate with a drug concentration of 80 μg/ml.The drug-containing solutions were incubated in a shaker at 37°C,sampling at 0,0.083,0.25,0.5,1,2,4,8,12,24,48 h.The samples were mixed with methanol at a ratio of 1:3,vortexes for 2 min,centrifuged at 13 000 rpm for 10 min,and the supernatant was taken for HPLC detection.
2.4. In situ single-pass intestinal perfusion study
All animal experiments followed the guidelines for animal use and were approved by the Ethics Committee of Shenyang Pharmaceutical University.The MES(4-Morpholineethanesulfonic acid hydrate)buffer was configured as reported before the experiment[20],and drugs were dissolved therein to have a drug concentration of 0.5 mM.In the experiment,Sprague-Dawley(SD)rats weighing 220-240 g were used.Previous studies verified the higher expression of MCT1 in the large intestine of human and rat through Western blot and immunostaining experiments.In particular,MCT1 is highly expressed in the distal colon,followed by the colon and ileum,and is rarely expressed in the jejunum and duodenum[17,21].After anesthesia with 20% urethane,the ileum and colon which with more MCT1 expression were found for cannulation perfusion experiments[17].The intestinal tract and the tube were saturated with blank MES buffer before administration for 20 min.The remaining inlet and effluent perfusate were collected and weighed at a predetermined time.The collected samples were mixed with methanol at a ratio of 1:3,vortexes for 2 min,centrifuged at 13 000 rpm for 10 min,and the supernatant was taken for HPLC detection.The absorption rate constantKaand the apparent permeability coefficientPappwere calculated by the following equation:
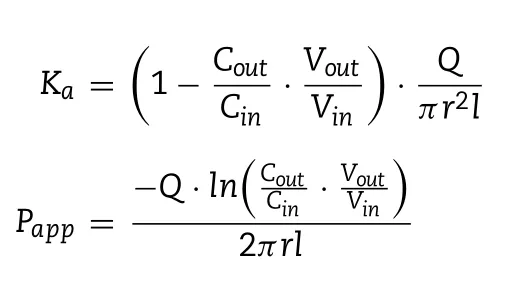
whereCinis the drug concentrations of the inlet perfusate andCoutis the drug concentrations of and outlet perfusate.VinandVoutare the volume of the inlet and outlet perfusate(the density of the perfusate is set to 1 kg/l),randlare the inner diameter(cm)and length(cm)of the intestine segment.Qis the flow rate of the perfusate.
2.5.Cell culture
The culture of Caco-2 cells was carried out using MEM-EBSS liquid medium.1% by volume of non-essential amino acids,1% sodium pyruvate,10% fetal bovine serum,100 U/ml penicillin and 100 U/ml streptomycin were added to the medium and stored at 4°C.The cells were cultured in a 5%carbon dioxide incubator at 37°C and saturated relative humidity.
2.6.Cytotoxicity
Caco-2 cells were plated in 96-well plate at 5×103cells per well.After 24 h of culture,the original medium was removed,and the drugs of different concentration gradients were added for further 48 h.After 48 h,the drug-containing solution was removed and added to a 20% MTT(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)medium,and cultured at 37°C for 4 h.The MTT solution was removed and added to 200 μl of DMSO(dimethyl sulfoxide)shakes for 15 min.The absorbance of the cell was measured at a wavelength of 490 nm to calculate IC50(half-inhibitory concentration).
2.7.Caco-2 cell uptake study
Caco-2 cells were plated in 24-well plate at 1×104cells per well.The liquid medium was changed every other day for the first 7 d,and the medium liquid was changed every day after 7 d until the 15th d of cultivation.In order to make the cells highly expressed MCT1,the medium containing butyric acid was replaced on the 7th d.The test was carried out 15 d later,the effects of time,drug concentration,and inhibitor on drug uptake were examined separately.The drug uptake experiments were carried out at a predetermined time and concentrations.The inhibitor concentration was 2.5 mM.After administration,the cells were cultured in a 37°C incubator,then the cells were washed rapidly with cold PBS solution for 3 times and collected with purified water and ultrasonic crushed.The concentration of the protein was determined by the bicinchoninic acid(BCA)method,and the concentration of the drugs was determined by the UPLC-MS/MS.
2.8.Caco-2 cell permeability
To evaluate the Caco-2 cell membrane permeability of the prodrugs,transwell experiments were performed.Caco-2 cells were plated in 12-well plate(Corning,Beijing,China)at 1×104cells per well.0.5 ml of cell-containing medium added in the chamber and 1.5 ml of blank medium at the bottom.The normal medium was changed every other day from the previous week,and then the medium containing butyric acid was changed until 21 d Caco-2 cells with good TEER values greater than 300Ωcm2were selected for prodrug transmembrane transport experiments.For the AL→BP experiment,washed the Caco-2 cell 3 times with 37°C HBSS buffer,then incubated in a 37°C incubator for 30 min.Then aspirated the Hank’s Balanced Salt Solution(HBSS)buffer and added moderate drug solutions and HBSS buffer to the intestinal side(AP side)and substrate side(BL side),respectively.The 12-well plate carrying the TranswellR○membrane was incubated in a constant temperature incubator.300 μl of HBSS buffer was taken from the BL side at 5,15,30,45,60,90,and 120 min,and 300 μl of blank HBSS buffer was added.For the BP→AL experiment,5-FU or prodrugs solutions were added to the basal side(BL side),and HBSS buffer was added to the intestinal side(AP side).The concentration of inhibitor was 2.5 mM.Samples were stored in a low temperature freezer at-80°C and tested by UPLC/MS/MS.Finally,the cell permeability results of the drug were calculated using the apparent permeability coefficient equation.
2.9.In vivo pharmacokinetics
Male Sprague-Dawley rats weighing 200-220 g were randomly divided into 5 groups(5 in each group),5-FU and prodrugs orally administrated at an equivalent 5-FU dose of 20 mg/kg,and one group intravenously administrated 5-FU at a dose of 8 mg/kg for calculation absolute bioavailability.The oral and intravenous administration groups had taken blood samples at a predetermined time.Blood samples were collected and placed in the-80°C refrigerator for testing.The plasma concentrations of prodrugs and 5-FU were simultaneously measured using UPLC-MS/MS.
Plasma samples were processed using the precipitated protein method.50 μl of plasma,50 μl of internal standard,100 μl of methanol were added to the eppendorf tube,vortexes for 3 min,and centrifugation at 13 000 rpm for 10 min.The supernatant was dried at 37°C under nitrogen,redissolved with methanol and water at a ratio of 15:85(v/v),vortexes for 3 min,centrifuged at 13 000 rpm for 10 min,and the supernatant was taken for injection.The compounds were well separated and quantified using an ACE UltraCore2 SuperC18(100 mm×2.1 mm id)silica gel column under the aqueous and acetonitrile gradient elution condition in an ACQUITY UPLC System(Waters).Ion transitions(MRM mode)werem/z145→42 for 5-CLU,m/z129→42 for 5-FU,m/z287→145 for HDA-5-FU.Calculation of pharmacokinetic parameters was performed by DAS software.
2.10.In vivo safety evaluation
Nine Kunming mice(male,18-22 g)were randomly divided into 3 groups.PBS,5-FU and OA-5-FU were administered intragastrically according to the dose of 40 mg/kg.Seven days after continuous intragastrically administration,the duodenum,jejunum,ileum,colon,heart,liver,spleen,lung and kidney of the mice were removed and placed in 10%formalin prepared for histological analysis.The immunochemical staining process was completed by steps such as embedding,dewaxing,dyeing,dehydration,and sealing to microscopic examination.
3.Results and discussion
3.1.Design and synthesis of prodrugs
HDA-5-FU and OA-5-FU were synthesized to target MCT1.HA-5-FU was also synthesized as a negative control.Briefly,a hydroxymethylation reaction occurred at the N1positions of 5-FU.In ice bath,carboxylic acid interacted with condensing agent HATU to form an active ester in the presence of TEA,and the active ester exchanged with 5-FU during the heating process to form a stable target product.The synthetic route and chemical structures were shown in Fig.1.Final products were mainly the single substitution product of the N1position,and there were also some N3mono-substituted and N1,N3disubstituted by-products.In order to obtain relatively pure product,products were separated by a preparative HPLC.Finally,all compounds were characterized by1H NMR,MS and HPLC(see the spectra information in Fig.S1-S9,Supporting Information).
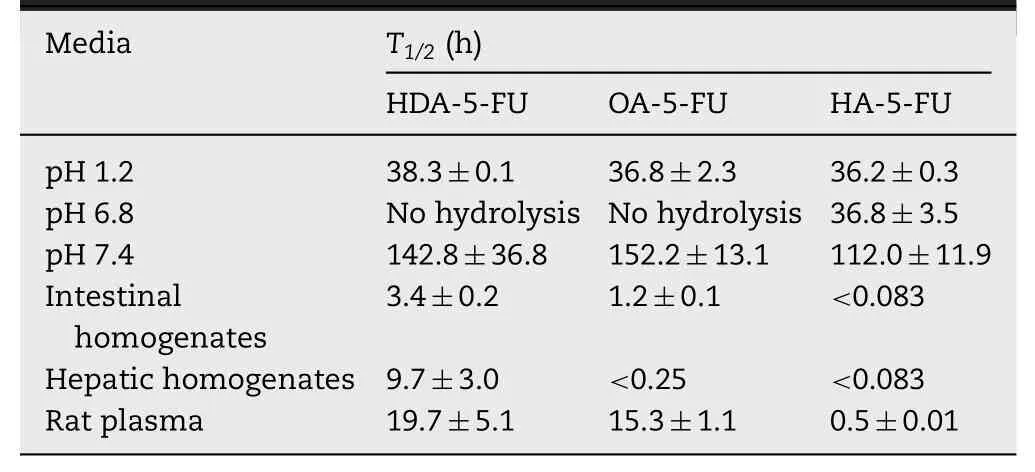
Table 1-Report on the stability of prodrugs in different pH solutions,rat tissue homogenates and plasma at 37°C.
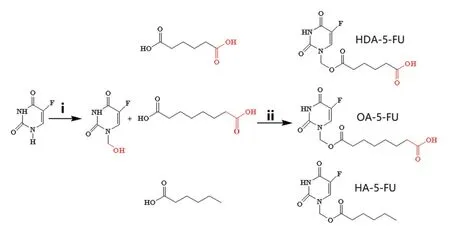
Fig.1-Synthetic scheme and chemical structures of 5-FU prodrugs.
3.2.Chemical and enzymatic stability evaluation
As a good prodrug for targeting intestinal transporters,it should have good gastrointestinal stability and appropriate bioactivation site.Therefore,stability studies were performed in different pH solutions,rat tissue homogenates and plasma.The half-life(T1/2)values were obtained by linear regression of pseudo-first-order kinetics(Table 1).As shown in Fig.2,prodrugs had good chemical stability in different solutions(pH 1.2,6.8 and 7.4)withT1/2>36 h,indicating that prodrugs was stable in GI tract and could be delivered in intact form,which was very important for transporter-targeting prodrugs.By contrast,it is shown in Table 1 that prodrugs were rapidly bioactivated in tissue homogenates and plasma,suggesting the prodrugs was stable in GI tract and then releases 5-FU after crossing intestinal membrane.Notably,stability of prodrug in intestinal homogenate indicates that a portion of prodrug was able to undergo biotransformation into 5-FU in intestinal tissue,which provides a basis for proper bioactivation of prodrugin vivo[22,23].
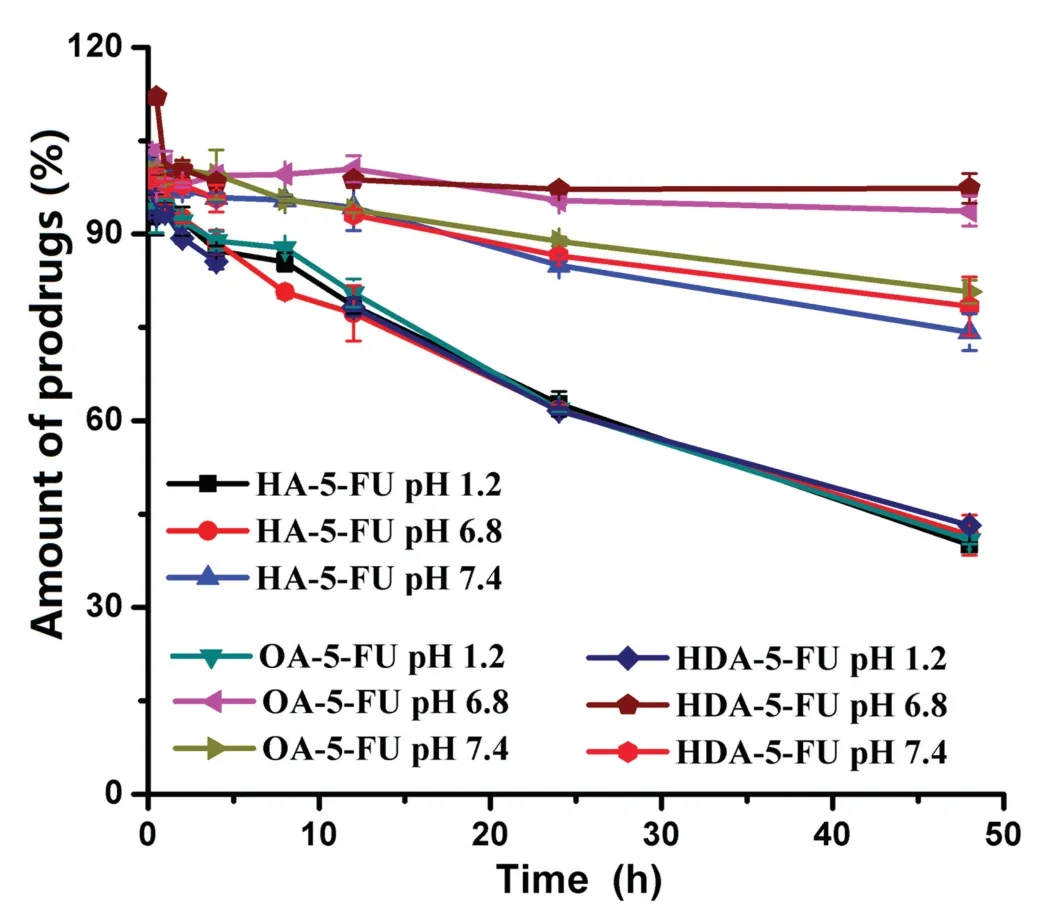
Fig.2-Stability of HA-5-FU(control),OA-5-FU(eight carbon linker)and HDA-5-FU(six carbon linker)in different pH solutions.
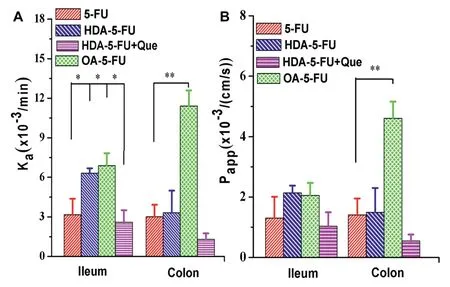
Fig.3-In situ absorption rate constant(Ka)(A)and apparent permeability coefficient(Papp)(B)of 5-FU,HDA-5-FU,OA-5-FU and HDA-5-FU addition with competitive inhibitor quercetin.Data are shown as mean±SD,n=3.∗P<0.05 versus 5-FU or inhibitor group and∗∗P<0.01 versus 5-FU or inhibitor group.
3.3. In situ single-pass intestinal perfusion study
To investigate the permeation and absorption of prodrugs,in situsingle-pass intestinal perfusion experiment was performed.Since MCT1 was expressed higher in the colon and ileum and had little expression in the duodenum and jejunum[17],the experiment was conducted only in colon and ileum.The modified 5-FU had better intestinal absorption and permeation than 5-FU.As shown in Fig.3,the absorption rate constant(Ka)and the apparent permeability coefficient(Papp)of OA-5-FU in the colon were 3.8-fold and 3.3-fold higher than5-FU,respectively.TheKaandPappof HDA-5-FU in the colon were 1.1-fold and 1.1-fold greater than 5-FU.These results illustrated that prodrugs could increase the absorption and permeation across intestinal epithelial cells.Quercetin is a class of competitive inhibitors but isn’t transported by MCT1.Currently,no reports have shown that quercetin binds to the hydrogen binding site on the MCT transporter.Therefore,it is possible that quercetin binds to the enzyme not at the active site,but outside the active site.MCT1-mediated transport could be inhibited by quercetin,which is competitive and reversible[24].To further demonstrate the increased permeability of prodrugs was due to the MCT1-targeting transport,MCT1 competitive inhibition studies were performed by using quercetin as MCT1 inhibitor[24].As shown in Fig.3,KaandPappvalues of HDA-5-FU were significantly decreased by co-incubating with quercetin,suggesting that the prodrugs could be the substrates of MCT1 transporter.Additionally,only a few amount of 5-FU was detected in the perfusion,indicating that MCT1-targeting oral prodrug was stable in GI tract.Because of rapid degradation of HA-5-FU in the perfusate,the intact prodrug form could not be detected and,thus,the results were no provided.
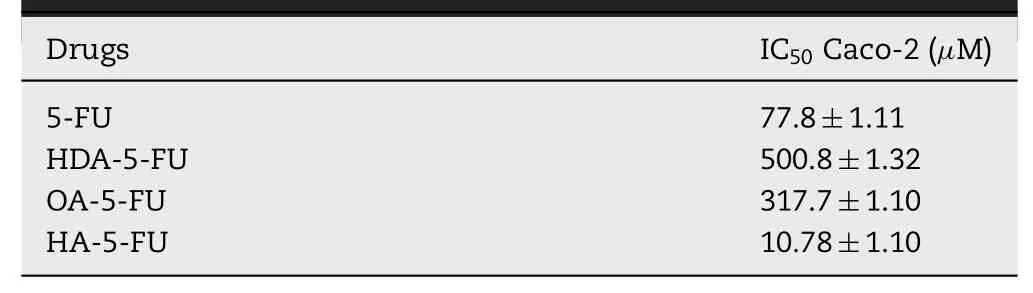
Table 2-In vitro cytotoxicity of 5-FU and prodrug to Caco-2 cell lines(MTT assay).
3.4.Cytotoxicity
In vitrocytotoxicity of 5-FU and prodrugs was evaluated in Caco-2 cells.As reflected in Table 2,IC50values in Caco-2 cells of HDA-5-FU and OA-5-FU were 6.4-fold and 4.1-fold greater than 5-FU.The MCT1-targeting prodrug was less effective in killing Caco-2 cells after incubation for 48 h,indicating the studied prodrugs had no toxicity in intestinal epithelial cells.On the contrary,IC50of HA-5-FU was relatively low,indicating HA-5-FU had potential gastrointestinal toxicity.The reason for this result may be due to the fact that HDA-5-FU and OA-5-FU were stable,while HA-5-FU was rapidly degraded and released 5-FU results in cytotoxicity.The prodrug is inactive and undergoes biotransformation to release an active parent drugs.In this article,the stability of HAD-5-FU and OA-5-FU was better than that of HA-5-FU,indicating that the biotransformation of HAD-5-FU and OA-5-FUin vivowas slower than that of HA-5-FU.Due to poor stability of HA-5-FU,it would be rapidly converted into 5-FU after entering cells,resulting in increased cytotoxicity[25].
3.5.Caco-2 cell uptake study
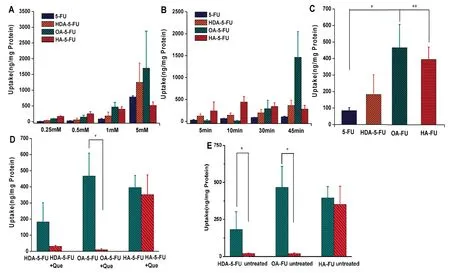
Fig.4-Uptake of 5-FU and prodrugs with increase of drug concentration(A)and time(B),respectively.(C)Uptake of drugs at the same uptake concentration and time(drug concentration is 1 mM and uptake time is 10 min).(D)Uptake of prodrugs addition with MCT1 competitive inhibitor.(E)Uptake of prodrug with/without treated by butyric acid.∗P<0.05 versus 5-FU or inhibitor group.∗∗P<0.01 versus 5-FU or inhibitor group.
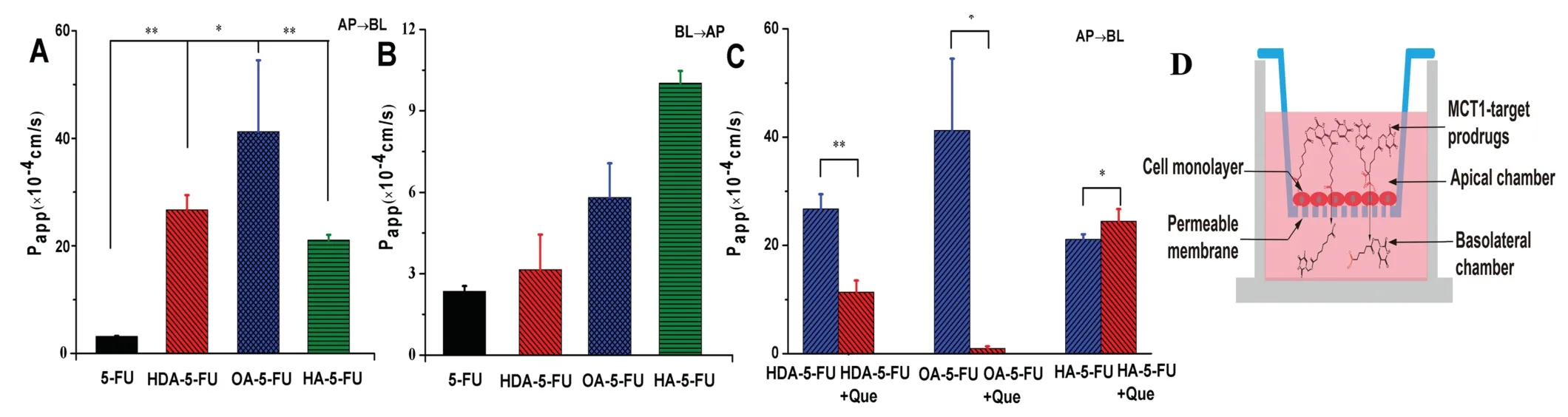
Fig.5-The transcellular results in Caco-2 cells.(A)The permeability of 5-FU and prodrugs from the apical side to the basolateral side.(B)The permeability of 5-FU and prodrugs from the basolateral side to the apical side.(C)The permeability of the prodrugs and 5-FU from the apical side to the basolateral side after addition with MCT1 competitive inhibitor.(D)Model of transport mechanism.∗P<0.05 versus 5-FU or inhibitor group.∗∗P<0.01 versus 5-FU or inhibitor group.
To affirm the contribution of MCT1 to the intracellular accumulation of the 5-FU prodrugs,the cellular uptake studies were investigated.The uptake amount of prodrugs and 5-FU increased with time and concentration(Fig.4A and B).The uptake of HDA-5-FU and HA-5-FU were 2.2-fold,and 4.7-fold higher than 5-FU,respectively(Fig.4C).Among the studied prodrugs,OA-5-FU exhibited the highest uptake,5.5-fold greater than 5-FU.These results indicated that MCT1 transporter contributed to the improved uptake of prodrugs and the different length of carbon chain could affect the uptake results of prodrugs.As seen in Fig.4D,the uptake of HDA-5-FU and OA-5-FU when co-incubating with MCT1 competitive inhibitor was 5.8-fold and 48.2-fold lower than those in the absence of inhibitor.However,for HA-5-FU,the addition of inhibitor had a little effect on its uptake.In addition,we conducted the uptake experiments on cells without induction.As shown in Fig.4E,the uptake of HDA-5-FU and OA-5-FU in untreated cells was significantly decreased,while the uptake of non-targeted prodrug HA-5-FU was not significantly changed.These results further proved that the monocarboxylic acid prodrugs could improve cellular uptake through MCT1 transporters.Notably,the cellular uptake of OA-5-FU was decreased mostly in the presence of competitive inhibitor,indicating the highest affinity with MCT1 transporters among the studied prodrugs.
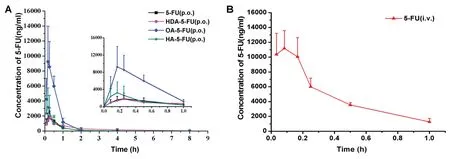
Fig.6-(A)Plasma concentration-time curve profile of 5-FU,HDA-5-FU,OA-5-FU,and HA-5-FU after oral administration with dose equivalent to 20 mg/kg 5-FU and(B)5-FU intravenous administration at a single dose of 8 mg/kg.The data is presented as means±SD(n=5).
3.6.Caco-2 cell permeability
The permeability result of 5-FU and prodrugs across a Caco-2 monolayer was shown in Fig.5.As seen from Fig.5,it could be observed that the membrane permeability of HDA-5-FU,OA-5-FU and HA-5-FU were 8.5-fold,13.1-fold and 6.7-fold greater than 5-FU during AP→BL transport.As for BL→AP transport,the membrane permeability of monocarboxylic acid prodrugs was not significantly different from that of 5-FU,suggesting MCT1 transporter was an absorptive transporter.The results of the inhibitor experiment showed that the membrane permeability of HDA-5-FU,OA-5-FU were 2.3-fold and 43.8-fold lower than those of 5-FU.By contrast,the nontargeted prodrug(HA-5-FU)wasn’t affected by MCT1 inhibitor,indicating that the membrane transport of HA-5-FU was mediated by passive diffusion,not by transporter-facilitating transport.In addition,the high permeability of OA-5-FU may be due to increased interaction of the side chain of octanedioic acid with the binding pocket of MCT1.
3.7.In vivo pharmacokinetics
The main purpose of designing prodrugs to target the intestinal MCT1 transporter was to improve the oral bioavailability.Therefore,pharmacokinetic experiments were performed at a single oral dose of 5-FU equivalent to 20 mg/kg in Sprague-Dawley rats.The plasma concentration-time curves of 5-FU and prodrugs(HDA-5-FU,OA-5-FU,HA-5-FU)were shown in Fig.6.A single dose of 8 mg/kg of 5-FU was administered intravenously in order to calculate absolute bioavailability.
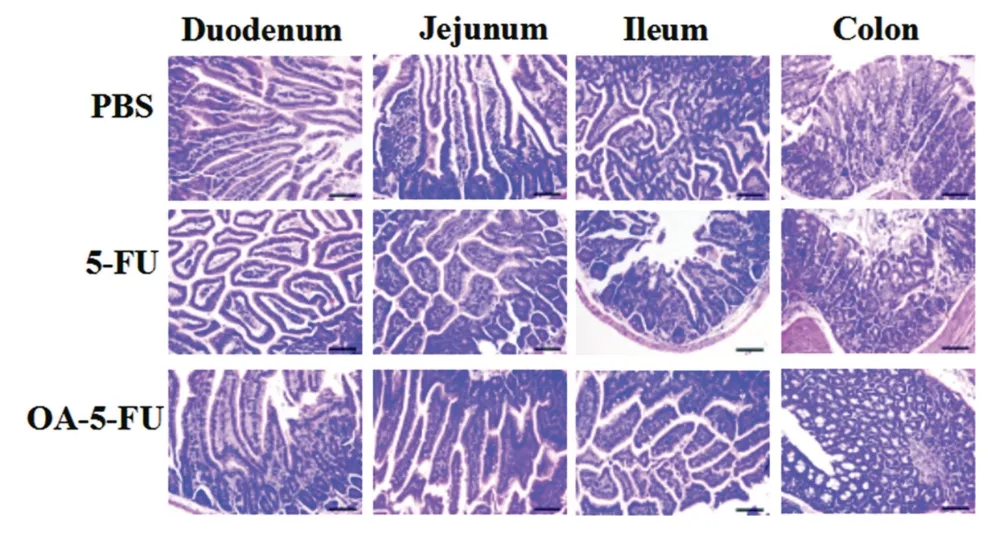
Fig.7-H&E staining results of duodenum,jejunum,ileum and colon of the three groups of Kunming mice after continuous administration for 7 d(ruler is 100 μm).
As shown in Table 3,Tmaxof 5-FU and its prodrugs after oral gavage was less than 20 min,indicating the prodrugs and 5-FU were rapidly absorbed from the intestine.The bioavailability of HDA-5-FU,OA-5-FU and HA-5-FU were 1.6-fold,4.1-fold and 1.3-fold higher than 5-FU.It was worth noting that OA-5-FU demonstrated the highest bioavailability due to good GI stability and permeability,and moderate bioactivation rate.The oral bioavailability of HDA-5-FU was relatively low because HDA-5-FU had low bioactivation rate.Moreover,the oral bioavailability of HA-5-FU was not significantly improved due to the poor stability of intestinal tissues.
3.8.In vivo safety evaluation
Poor selectivity of 5-FU might lead to severe intestinal toxicity after oral administration.In contrast,inactive prodrugs might have low intestinal toxicity.Therefore,in vivosafety assessment was performed to verify the hypothesis.The duodenum,jejunum,ileum and colon of the administered mice were isolated and histologically stained.In the 5-FU group(Fig.7),inflammatory cell infiltration occurred in the duodenum,jejunum and ileum tissues and a small amount of inflammatory cells infiltrated in the colon tissue,and the intestinal villi became shorter.No significant toxicity was observed in the small intestine of the PBS-treated and OA-5-FU-treated groups,probably because of the prodrug existed as an intact form in GI tract and had not been degraded into toxic 5-FU.To verify the systemic safety,we conducted histological studies on the heart,liver,spleen,lung and kidney in Kunming mice.In histological studies,the drug dose was two-fold higher than that in pharmacokinetics.Seven days after continuous intragastrical administration,the liver cells were wrinkled,the nuclei were pyknotic,a small amount of blood could be seen between the liver plates,the pulmonary septum was thickened significantly in the group of 5-FU,and the renal tubular epithelial cells were significantly swollen.The enterocytes in the prodrugs and PBS groups were relatively intact,and the tissues showed no significant changes.This further demonstrated the systemic safety of the prodrugs and verified that there was no systemic toxicity of the prodrugs after continuous intragastrical administration.This result shown that the prodrug OA-5-FU could reduce the toxicity of 5-FU and improved the safety of 5-FUin vivo(Fig.8).
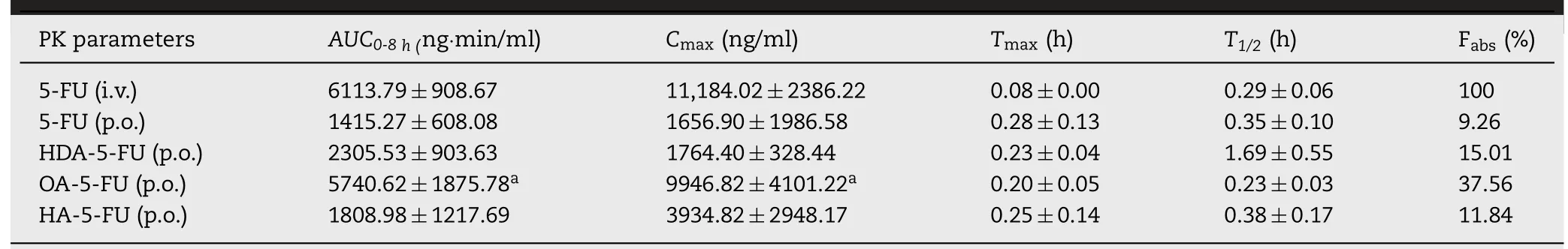
Table 3-Pharmacokinetic parameters of 5-FU intravenous administration and 5-FU,HDA-5-FU,OA-5-FU and HA-5-FU oral administration.The parameters are expressed as a mean±SD(n=5).
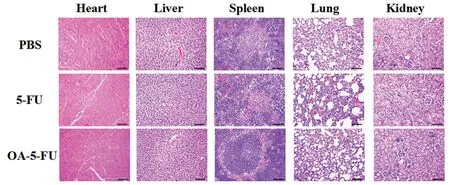
Fig.8-H&E staining results of heart,liver,spleen,lung,and kidney of the three groups of Kunming mice after continuous administration for 7 d(ruler is 100 μm).
4.Conclusions
In this study,MCT1-targeted oral prodrugs were designed for 5-FU.Two MCT1 targeting conjugates were developed to improve oral delivery of 5-FU by simultaneously modulating GI stability,membrane permeability andin vivobioactivation.Moreover,OA-5-FU and HDA-5-FU were adequately proven to be typical substrates of MCT1 by means of Caco-2 cells studies.The membrane permeability(13.1-fold)and oral bioavailability(4.1-fold)of OA-5-FU were increased greatly,in comparison with 5-FU and its non-targeted prodrug counterpart HA-5-FU.These results indicated that good targeting and appropriate bioactivation rate were significant factors in the design of oral prodrug.The low gastrointestinal toxicity of OA-5-FU alleviate the occurrence of side effects of 5-FU.In summary,the intestinal MCT1 transporter was a promising intestinal target for exploiting oral prodrugs.Good gastrointestinal stability,high membrane permeability and appropriate bioactivation were the three important aspects to design high-efficiency oral prodrugs targeting intestinal transporters.
Conflicts of interest
The authors declare that there is no conflicts of interest.
Acknowledgments
This work was financially Supported by National Nature Science Foundation of China(No.81773656,U1608283,81573497),Liaoning Revitalization Talents Program,No XLYC1808017,Key projects of Technology bureau in Shenyang,No18400408,Key projects of Liaoning Province Department of Education,No.2017LZD03.
Supplementary material
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2019.04.001.
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Cancer nanotechnology:Enhancing tumor cell response to chemotherapy for hepatocellular carcinoma therapy
- The functions and applications of A7R in anti-angiogenic therapy,imaging and drug delivery systems
- Investigation of molecular aggregation mechanism of glipizide/cyclodextrin complexation by combined experimental and molecular modeling approaches
- Evaluation of the Mrp2-mediated flavonoid-drug interaction potential of quercetin in rats and in vitro models
- Synthesis,characterization and in vivo evaluation of honokiol bisphosphate prodrugs protects against rats’brain ischemia-reperfusion injury
- Improved dissolution and oral absorption by co-grinding active drug probucol and ternary stabilizers mixtures with planetary beads-milling method