Investigation of molecular aggregation mechanism of glipizide/cyclodextrin complexation by combined experimental and molecular modeling approaches
2020-01-06
State Key Laboratory of Quality Research in Chinese Medicine,Institute of Chinese Medical Sciences(ICMS),University of Macau,Macau,China
ABSTRACT Cyclodextrin complexation is a wise strategy to enhance aqueous solubility of waterinsoluble drugs.However,the aggregation mechanism of drug-cyclodextrin complexes is still unclear.This research aimed to investigate the molecular aggregation mechanism of glipizide/cyclodextrin complexation by the combination of experimental and modeling methods.Binding free energies between glipizide and cyclodextrins from modeling calculations were higher than those by the phase solubility diagram method.Both experimental and modeling results showed that methylated-β-cyclodextrin exhibited the best solubilizing capability to glipizide.Size-measurement results confirmed the aggregation between glipizide and all four cyclodextrins in high concentrations.Glipizide/γ-cyclodextrin and glipizide/β-cyclodextrin complexes showed stronger aggregation trend than HPβ-cyclodextrin and methylated-β-cyclodextrin.The substituted groups in the rim of HP-β-cyclodextrin and methylated-β-cyclodextrin lead to weak aggregation.This research provided us a clear molecular mechanism of glipizide/cyclodextrin complexation and aggregation.This research will also benefit the formulation development of cyclodextrin solubilization.
Keywords:Glipizide Cyclodextrin Molecular modeling Aggregation Molecular mechanism
1.Introduction
Cyclodextrins(CDs),as one type of pharmaceutical excipients,are widely used to improve solubility and dissolution rate of water-insoluble drugs[1-3].CDs can form complexes with drugs because of its hydrophobic cavity and hydrophilic shell[4-9].The cavities of CDs could encapsulate unstable or distasteful drugs and this application has already been widely used[10].The aqueous solubility of the natural CDs(e.g.β-CD)is much lower than that of other acyclic saccharides because of their strong inter-molecular hydrogen bonding in the crystal state.Hydroxyl or methyl substitutions groups result in significant improvement of their aqueous solubility.The hydroxypropyl-β-CD(HP-β-CD)and the methylated-β-CD(Me-β-CD)are most commonly used[1,11].Usually,the binding free energy of inclusion complexes is determined by phase-solubility diagrams[12,13].Molecular dynamics simulation is also widely used to assess the steric interaction of complexes and predict the characteristic of drug-CD interaction in either vacuum or solutions[14-18].However,the individual drug/CD complex can only exist in ideal dilute solutions[19].According to the literature reported[20],CDs were able to form both inclusion and non-inclusion complexes.In the real solution,there were many different types of solutes,such as free drug molecules,“empty”CD molecules,drug/CD inclusion complexes and drug/CD non-inclusion complexes[21].Several reports had indicated that two or more CDs and CD complexes can self-associate to form aggregates[19,22-30].These aggregates were able to enhance the solubility of drugs through non-inclusion complexes or“micelle-like”structure.The formation of hydrogen bonds between natural CDs and water lead to the aggregation,while the substituted groups in the rim of HP-β-CD and Me-β-CD hinder the selfaggregation[19].Moreover,the particle of aggregates was too small to be seen.For example,2,5-Diphenyloxazone-γ-CD was found to form aggregates in its high-concentration solutions[26].The formation ofγ-CD aggregates in aqueous solution and their critical aggregation concentration were determined by both conventional dynamic light scattering(DLS)method and new permeation method for quantitative determination ofγ-CD[31].The drug/γ-CD/HP-γ-CD complex was stabilized by poloxamer-407 through the formation of multi-component aggregates[32].However,the importance of CD aggregates was still underestimated by academic and industrial scientists.
Glipizide(GLI),a second generation sulfonylurea commonly prescribing drug[33],is used to treat type II diabetes mellitus.GLI can efficaciously reduce the blood glucose level,metabolism and delay the occurrence of vascular complications in diabetic patients.GLI is a weakly acid drug with poor water-solubility at room temperature.Its poor aqueous solubility limits its absorption in gastrointestinal(GI)tract[34-37].It has been reportedβ-CD,HP-β-CD and Me-β-CD have the good solubilizing effect on GLI[3,34,38-45].For instance,GLI/β-CD complexes were prepared for controlled release matrix tablets,which showed better solubility and stability than pure GLI[3].GLI was used as a model drug to evaluate a newly modified CD and compared with GLI/HP-β-CD complexes,the formation of GLI/CD complexes showed better solubilizing effect and the newly modified CD showed better solubility effect to GLI than HP-β-CD[41].GLI/β-CD complexes were compared with GLI/Me-β-CD complexes in phosphate buffer(pH=4 and pH=8).It suggested the encapsulation of glipizide withβ-CD and its methyl and hydroxypropyl derivatives was a useful strategy to improve the solubility and dissolution of these poorly soluble therapeutic agents and Me-β-CD had the better solubility-enhancement ability to glipizide[38].Previous works mainly emphasized on the formation and properties of the inclusion complexes.
The aim of this study was to investigate the molecular aggregation mechanism of the drug/CD complexes by both experimental and modeling approaches.GLI was used as the model drug and four CDs(β-CD,γ-CD,HP-β-CD,Me-β-CD)which are widely used in the commercial and experimental field were selected.Phase solubility studies,Fourier transform infrared spectroscopy(FTIR)studies,differential scanning calorimetry(DSC)studies and molecular dynamic simulation of 1:1 GLI/CD complexes were used to investigate binding free energies and complexation mechanisms.Dynamic light scattering(DLS)studies and MD simulation of 10:10 GLI/CD aggregates were used to investigate aggregation size and mechanisms.
2.Materials and method
2.1.Materials
Glipizide was purchased from Wuhan Dongkang Technology Company Limited(China).β-CD,γ-CD,HP-β-CD and Me-β-CD were purchased from Beijing J&K Scientific Company Limited(Beijing,China).Methanol was purchased from MerkKGaA Company(China).Ethanol absolute was purchased from Tianjin Damao Chemical Reagent Factory(Tianjin,China),China.All reagents and solvents were of analytical grade.
2.2.Binding free energy calculation by phase solubility studies
Phase solubility studies were referred to methods of previous research[13,46].In brief,excessive amounts of GLI were dissolved in distilled water containing various concentrations of CDs and placed in a water bath oscillator for 2 d under different temperature.Subsequently,samples were filtered by 0.45 μm filter membrane.The filtered samples were diluted suitably and analyzed by DR6000 Ultraviolet-visible(UV-Vis)Spectrophotometer Hach at 275 nm.Each sample was tested three times.The inclusion constant(Kc)was calculated by the formula:Kc=Slope/S0×(1-Slope).S0was the intrinsic solubility of GLI.
2.3.Preparation of inclusion compound
The solid inclusion complex was prepared with saturated water solution method.The solid inclusion complexes were prepared at 1:40 weight ratio of GLI to CDs.100 mg GLI were dissolved in 100 ml methanol solution.The GLI methanol solution was added to the CD solution(0.015 mmol/ml forβ-CD and 0.05 mmol/ml for other CDs),dropwise with slow stirring for 2 h under 60°C.Then,the mixtures were stirred at room temperature for 4 h.The suspension was stored under 4°C overnight.After filtration,the sediment was washed with acetone for 2-3 times.The complexes were dried in the oven and was sieved through 80 mesh for further analysis.
2.4.Characteristic of GLI/CD complexes by FTIR studies
The GLI/CD complexes prepared by saturated water solution method were recorded on Thermo Nicoletis 10 IR using KBr disc.The FTIR results of GLI/CD complexes were compared with those of GLI,CD,and GLI/CD physical mixture.The data was analyzed under OMINIC program.Physical mixtures were prepared at the same weight ratio as the complexes.
2.5.Characteristic of GLI/CD complexes by DSC studies
The DSC measurement of GLI,CD,GLI/CD physical mixtures,and GLI/CD complexes was performed using Shimadzu Different Scanning Calorimetry(DSC)-60A Systems(Shimadzu,Kyoto,Japan)and equipped with Thermal Analysis(TA)-60 workstations.The thermal behaviors were studied by heating all samples in a sealed aluminum pan with an empty sealed pan as a reference.The temperature range was 30-250°C with a rate of 10°C/min under a nitrogen gas stream.
2.6.Size measurement of GLI/CD solution by DLS method
The particle size characteristics were performed by DLS method under ZETA-SIZER NANO ZSP.An excessive amount of GLI was dissolved in distilled water containing 0.02 mmol/ml concentration of CDs(the concentration ofβ-CD was 0.015 mmol/ml because of its saturated concentration)and placed in a full temperature shock incubator for 2 d.Subsequently,the samples were filtered.Measurements were carried out at a scattering angle of 173° under ZETA-SIZER NANO ZSP software system.The size distributions were obtained from intensity autocorrelation function by regularization analysis.The selections of filter membranes and other parameters were the same as previous research[25].Free CDs and GLI solutions under same concentration were tested as a blank control.Each measurement was done in triplicate.
2.7.Simulation details
2.7.1.Molecular structures construction
The structures ofβ-CD andγ-CD were obtained from the Cambridge Crystallographic Data Center[47],HP-β-CD and Me-β-CD were modified from the structure ofβ-CD by Discovery Studio 2016 Client.The position of substituent was referred to previous paper[30,48].GLI was drowned by Discovery Studio 2016 Client according to Chinese pharmacopoeia 2015.All the molecular structures were optimized with a force field by Discovery Studio 2016 Client.
2.7.2.The construction of initial structures of 1:1 GLI/CD simulation by docking
AutoDock Vina is widely used to predict the affinity of 1:1 ligand-receptor interaction[49,50].The AutoDock Tools package and AutoDock Vina were used to perform docking studies.GLI and CD were combined by the ratio of 1:1[34,45].In docking simulation,the CD was a“receptor”while GLI was a“ligand”.Semi-flexible docking method was adapted to GLI.Docking coordinates and center of the search space are shown in Table 1.All the other parameters were default values in AutoDock Vina.The structures with the lowest affinity would be considered as stable binding models and performed in MD simulations as initial structures.
2.7.3.One-to-one molecular dynamics(MD)simulation
The initial structures of one-to-one(1:1)MD simulation were obtained from Section 2.7.2.The MD simulation was carried out under the AMBER 14 and AMBER Tools 14 software package.The general AMBER force field was applied in the antechamber module for all molecules.The transferable intermolecular potential with 3 points(TIP3P)water model with a cube of 20 ˚A thicknesses was added to solvate each system.Table 2 showed the system construction.During the MD simulation of each 1:1 GLI/CD system,the solvent was firstly subjected 10 000 steps of minimization of solvation energy and the whole system has conducted a total of 20 000 steps of the minimization.The system was heated to 37°C in a total of 10 000 steps MD simulation after energy minimizations.After heating,70 ns MD simulation was performed with a time step of 0.002 ps and a cut-off of 10 ˚A.
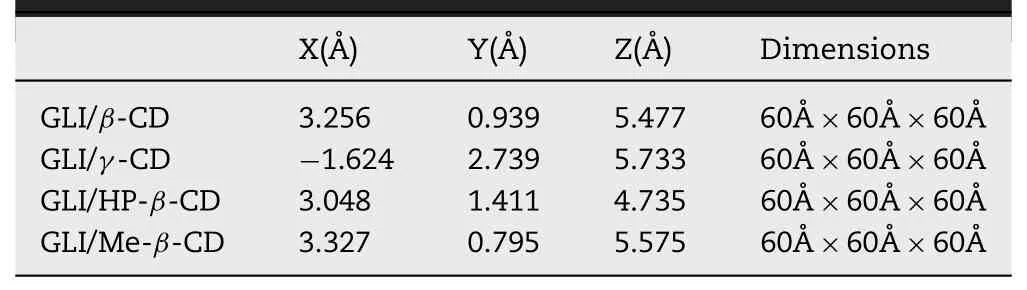
Table 1-Docking coordinates and center of the search space in docking simulation.
2.7.4.Binding free energy calculation by molecular mechanics/Poisson-Boltzmann surface area(MM/PBSA)method
The MM/PBSA method is widely used to calculate the binding free energy of 1:1 ligand-receptor system[51].The average interaction energies of the receptor and the ligand were calculated by the last 5 ns structures from the MD trajectory.The binding free energy of the GLI/CD inclusion complex(ΔGbind)was calculated by the free energy of complex(ΔGcomplex)and the isolated GLI(ΔGGLI)and CDs(ΔGCD)as the following equation:
ΔGbind=ΔGcomplex-ΔGGLI-ΔGCD
The absolute molecular mechanical energy changes(ΔEMM)were calculated with the following equation:
ΔEMM(totalgasphaseenergy)=ΔEinternal+ΔEvdw+ΔEelectrostatic
ΔEvdwandΔEelectrostaticwere the electrostatic and Van der Waals interaction energy changes,the internal energy changes(ΔEinternal)represented the strain energy changes in bonds,angles and torsion angles.
The Gibbs free energy(△G)was calculated by the enthalpy(△H)and entropy(△S)with invariable temperature(T):
ΔG=ΔH-TΔS
2.7.5.Multi-to-multi MD simulation
In the multi-to-multi(10:10)MD simulation part,the initial structures of MD simulation were built by“PACKMOL”package with the tolerance distance 3.0 ˚A and 100 ˚A×100 ˚A×100 ˚A cube.10 GLI and 10 CDs were performed.14 ˚A thickness TIP3P water was added in each system.Table 3 showed the system constructions of 10:10 systems.10:10 GLI/CD systems were simulated by 150 ns.Other settings were the same as Section 2.7.3.

Table 2-1:1 system constructions and information of molecular number and atom number.
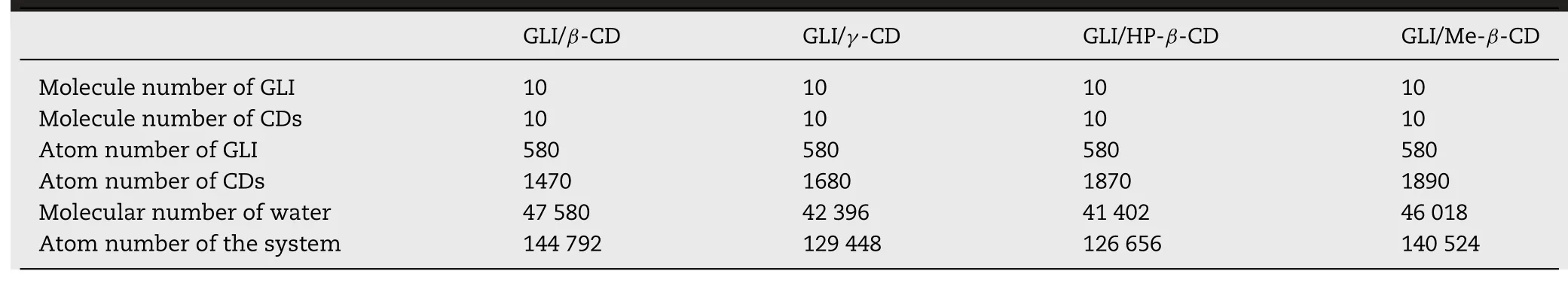
Table 3-10:10 system constructions and information of molecular number and atom number.
2.7.6.Multi-to-multi system analysis
CPPTRAJ is the main program in AMBER for processing coordinate trajectories and data files.Root-mean-squared deviation(RMSD)of mass,the solvent-accessible surface area(SASA)of GLI and CDs,number of contact points within 3 ˚A between GLI and CDs,number of water molecules within 3.6 ˚A around GLI and hydrogen bonds information were selected as the evaluation parameters.All the parameters referred to AMBER 2017 manual and previous paper[52].RMSD coordinates were calculated by the reference to the initial structures.The solvent-accessible surface area used the“linear combination of pair-wise overlap”(LCPO)algorithm of Weiser et al.[53].Any atoms between GLI and CDs which were closer than 3 ˚A in the specified reference frames were considered as native contact points and these atoms reflected aggregation[54,55].Number of water molecules around GLI within 3.6 ˚A reflected the environment around drugs.All the data were performed under CPPTRAJ program and exhibited in XMGRACE package.
3.Results and discussion
3.1.Results and discussion of 1:1 GLI/CD complexes
3.1.1.Binding free energy calculation by phase solubility studies
Solubility experiments were carried out to investigate interactions between GLI and CDs.The phase solubility diagrams of GLI/CD solution under 37°C are shown in Fig.1.
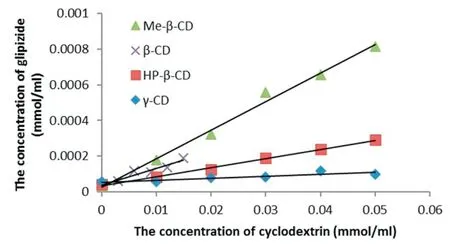
Fig.1-Phase solubility diagrams of GLI with four CDs in distilled water at 37°C.
The phase solubility diagram of GLI/CD could be classified as ALtype according to Higuchi and Cornnors which confirmed the stoichiometry of 1:1 complex[13].The intrinsic solubility of GLI was 2.70±0.97×10-5mol/ml under room temperature and 3.93±0.86×10-5mol/ml under 37°C which agreed with those of previous paper(2.29×10-5mol/ml in room temperature and 4.08×10-5mol/ml in 37°C)[34,41,45].The aqueous solubility of the GLI increased linearly as a function of the concentration of CDs.Table 4 showed the binding constant(Kc)under different temperature,enthalpy(ΔH),entropy(ΔS)and binding free energy(ΔG)calculated from the formula:ΔG=-R×T×ln(kc)and ΔG=ΔH-T×ΔS.Negative enthalpy indicated the GLI/CD complexation reaction was exothermic.It has been reported that ΔG of GLI/β-CD and GLI/Me-β-CD complexes was-17.71 kJ/mol and-18.27 kJ/mol under 37°C and those of ours were-14.02 kJ/mol and-15.52 kJ/mol[34,38].These differences were produced because the previous experiments were carried out under different pH and CD concentrations.According to the literature reported,the Kc of GLI/HP-β-CD complexes was 286 M-1under 30°C as well as ΔH and ΔS was-57.11 kJ/mol and-142.31 J/mol/K under the similar experimental condition as ours.These results were in accordance with our experimental results.Kc decreased with the increased temperature which proved that heating was not benefited to the complexation.GLI/HP-β-CD had the lowest entropy because the intramolecular hydrogen bonds of HP-β-CD limited the free-moving of GLI in the CD cavity which has been explained in the previous research[56].
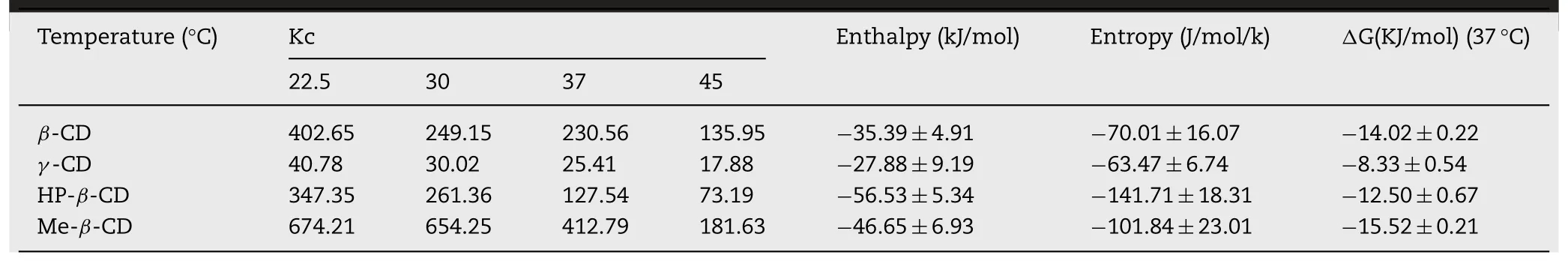
Table 4-Binding constant(Kc),enthalpy and entropy of GLI in CDs solution under different temperature.
According to phase solubility results,the solubilization capability of GLI by CDs was as follows:Me-β-CD>β-CD>HP-β-CD>γ-CD under same experimental conditions.
3.1.2.Characteristic of GLI/CD complexes by FTIR studies
The FTIR results gave references to the 1:1 modeling results,the substituent groups of GLI wrapped in CD could be observed by combined FT-IR and modeling investigation.
GLI,CDs,GLI/CD physical mixture and GLI/CD complexes were analyzed under OMINIC program.The IR spectra of GLI,CD,physical mixture(pm)and GLI/CD complexes(com)are shown in Fig.2.The variation of the shape,shift,and intensity of the IR absorption peaks of GLI and CDs could provide enough information for the occurrence of the inclusion.GLI had a double characteristic peak in 3326 cm-1and 3251 cm-1which represented N-H stretching.The double peak in 1690 cm-1and 1651 cm-1represented C=O stretching,the double peak of 1160 cm-1and 1035 cm-1represented S=O stretching in GLI.Aromatic vibration had a single peak in 1529 cm-1in GLI and C-H of cyclohexane bending had a peak in 1445 cm-1in GLI.According to FTIR results ofβ-CD/GLI complexes,3382 cm-1was the characteristic peak of-OH,2926 cm-1was the characteristic peak of anti-symmetric stretching vibration peak of CH2,1028 cm-1was the characteristic peak of C-O-C and 1157 cm-1was the characteristic peak of C-O stretching peak.The spike of physical mixture was the combination of GLI and CDs.The major characteristic peaks of GLI remained in physical mixture and the intensity significantly reduced.The disappearance of some GLI characteristic peaks in GLI/β-CD complexes indicated the existence of GLI/β-CD complexes.The similar conclusion could also be obtained from other FTIR results.
According to the FTIR results,it could confirm the disappeared characteristic peak of GLI in the complexes,which could confirm the formation of complexes.
3.1.3.Characteristic of GLI/CD complexes by DSC studies
DSC results confirm the formation of complexes which could also be compared with final structures of modeling.
DSC curves of pure GLI,CDs,GLI/CD physical mixture and GLI/CD complexes are shown in Fig.3.GLI had a single sharp peak at 208.6°C which corresponded to the melting point.β-CD had a broad endothermic peak which indicated the evaporation of water.The DSC curve of GLI/β-CD physical mixture was the superposition of the individual components.The thermal characteristic peak of drug was clearly distinguishable in the physical mixture and its intensity reduced.These little changes suggested a weak interaction between GLI andβ-CD during the mixing or heating for DSC scanning.However,the complete disappearance of the endothermic peak of GLI in the GLI/β-CD complexes suggested that GLI was well inserted in theβ-CD cavity.The other DSC curves of GLI/CD showed a similar phenomenon which could also prove the formation of GLI/CD complexes.
3.1.4.MD simulations of 1:1 GLI/CD system and the comparation of experimental and modeling results
MD simulation of 1:1 GLI/CD complexes could mimic the GLI/CD complexes in ideal dilute solution.
Initial structures of one-to-one MD simulation were obtained from AutoDock Vina.Fig.4 showed the final stable structures after 70 ns MD simulation.β-CD wrapped the cyclohexane and N-H characteristic groups of GLI.γ-CD wrapped cyclohexane,benzene and C=O characteristic groups of GLI.HP-β-CD and Me-β-CD wrapped the benzene ring,N-H and S=O of GLI.The groups wrapped by CDs were consistent with the disappeared characteristic peaks in FTIR.
The phase solubility results and binding free energy calculated by MM/PBSA could be compared.Table 5 showed the binding free energy results of 1:1 GLI/CD complexes by MM/PBSA method.GLI was partially entered the cavity of CD,which limited their thermal motion and resulted in large changes of enthalpy.The favorable enthalpy fetched up the unfavorable enthalpy.△G was negative which indicated that the reactions could proceed spontaneously.Negative△H,which agreed with experimental results,proved that the reaction released heat.△S were negative which suggested the reaction was the process of entropy reducing[57].The solubilization effect of CDs to GLI investigated by MM/PBSA method was:Me-β-CD>γ-CD>β-CD>HP-β-CD.
According to experiments and 1:1 modeling studies,structures and binding free energies of GLI/CD complexes had been investigated.GLI was wrapped within the cavity of CD molecule.The disappeared FTIR characteristic peaks and melting point of GLI in the complexes confirmed the formation of complexes.Both phase solubility and modeling studies suggested that Me-β-CD had the best solubility enhancement capability to GLI.All binding free energies from 1:1 modeling studies showed higher values than those of experimental data,especially GLI/γ-CD system.Our previous research also showed a similar situation[47].The possible reason for the difference was that the experimental data came from high-concentrated solution,but the 1:1 modeling only simulated the ideal dilute solution.Therefore,1:1 systems can’t fully reflect the real experiments of drug/CD complexation well.A better model should be developed to shed light on the complexes mechanism involving in both inclusion and non-inclusion complexations.

Fig.2-FTIR spectra of pure(black)CDs(A.β-CD B.γ-CD C.HP-β-CD D.Me-β-CD),(red)GLI,(blue)GLI/CD physical mixtures and(green)GLI/CD complexes.(For interpretation of the references to color in this figure legend,the reader is referred to the web version of this article.)
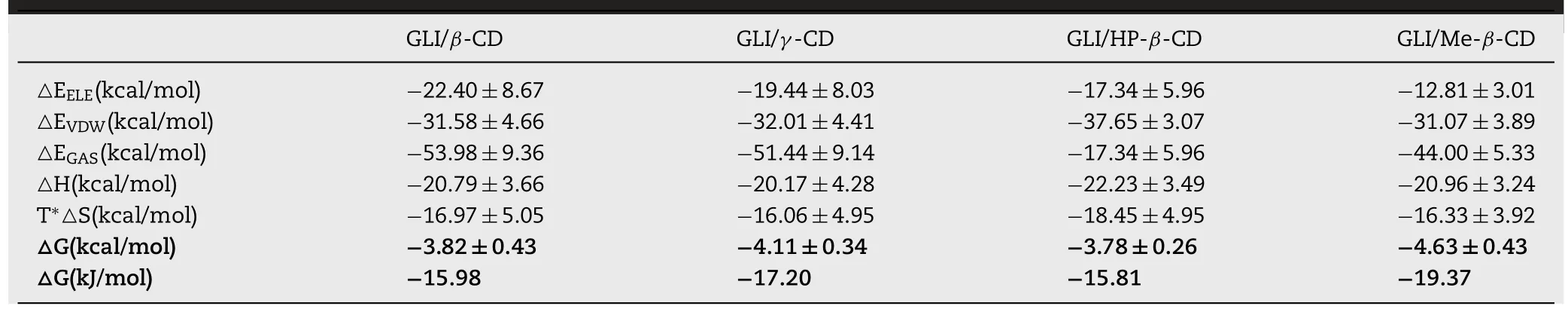
Table 5-Binding free energy and energy components under MM-PBSA calculation.
3.2.Results and discussion of GLI/CD aggregates
3.2.1.Size-measurement of GLI/CD solution by DLS method
The DLS results could be compared with 10:10 modeling results to confirm the aggregation capability of CDs.
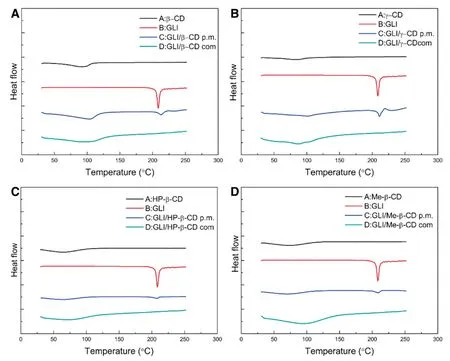
Fig.3-DSC thermograms of pure(black)CDs(A.β-CD B.γ-CD C.HP-β-CD D.Me-β-CD),(red)GLI,(blue)GLI/CD physical mixtures and(green)GLI/CD complexes.(For interpretation of the references to color in this figure legend,the reader is referred to the web version of this article.)
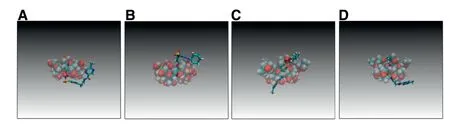
Fig.4-Snapshots of final structure of 1:1 GLI/CD simulation.Carbon elements are indicated in blue bonds,hydrogen elements in white,and oxygen elements in red.(A)GLI/β-CD complexes,(B)GLI/γ-CD complexes,(C)GLI/HP-β-CD complexes,(D)GLI/Me-β-CD complexes.(For interpretation of the references to color in this figure legend,the reader is referred to the web version of this article.)

Table 6-Mean hydrodynamic radii and polydispersities(in nm)of GLI in different concentration of CDs through 0.22 μm filter.
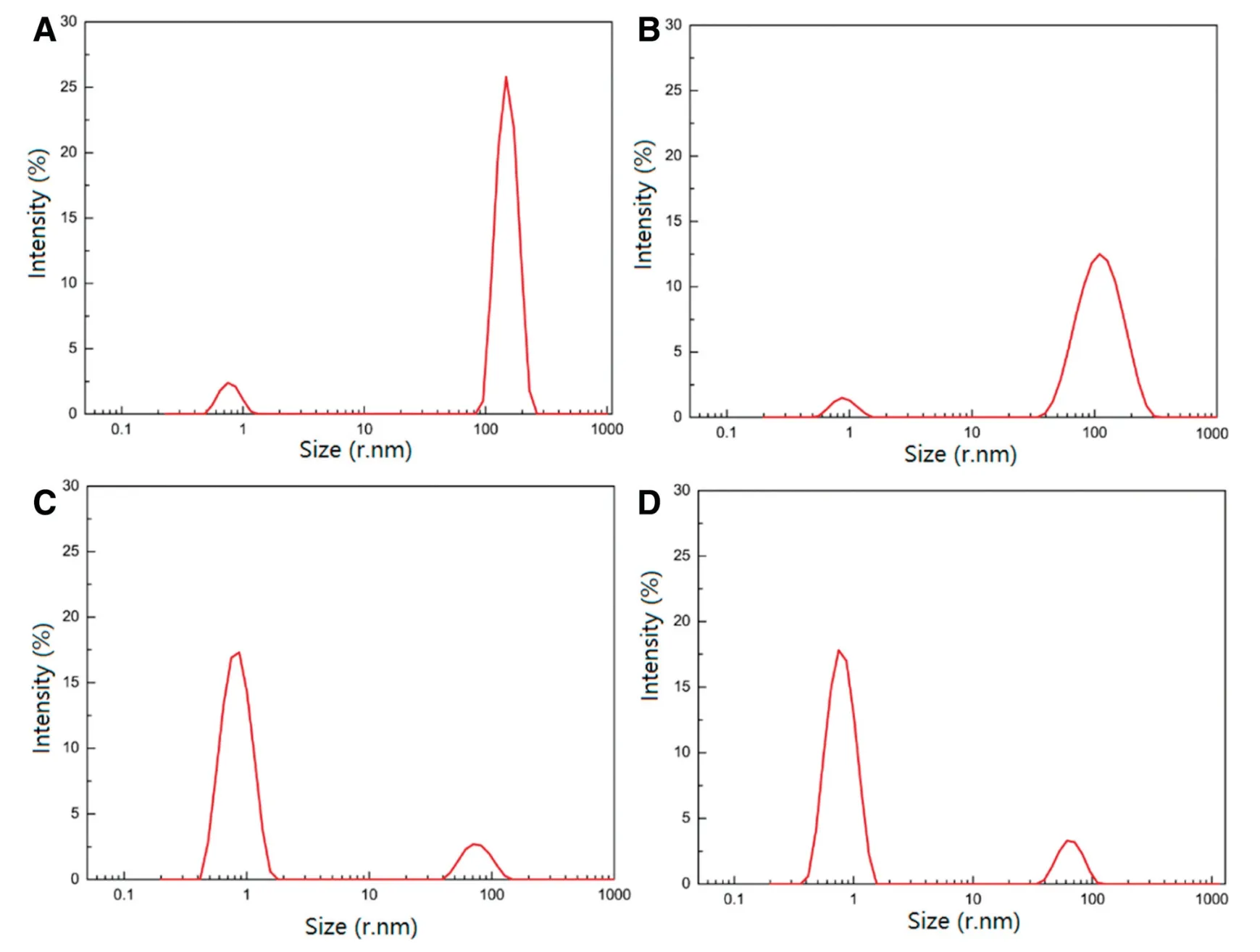
Fig.5-DLS correlogram regularization analysis for GLI/CD solution.(A)GLI/β-CD solution,(B)GLI/γ-CD solution,(C)GLI/HP-β-CD solution,(D)GLI/Me-β-CD solution.
According to Table 6 and Fig.5,a bimodal distribution is evident from each result.The DLS measurement couldn’t detect a significant peak in the saturated solution of GLI possibly due to the low intrinsic solubility of GLI.The first peak has a lower radius corresponding to monomeric GLI/CD complexes.The second peak however,has a higher radius which is attributed to aggregates.From the results,the hydrodynamic radii for the monomers are close to the actual dimensions of GLI/CD complexes.The sizes of freeβ-CD andγ-CD aggregates were 141.70±18.62 nm and 95.89±43.97 nm respectively.Free HP-β-CD and Me-β-CD aggregates were dominantly monomers and had very low intensity.As stated in the previous research on this topic,the aggregation size of CDs under certain conditions is usually less than 300 nm.This finding concurs with our results[22,25,58,59].Fig.5 showed the observation made on the distribution of polydisperse mode,the mean hydrodynamic radius(percentage of intensity)of GLI/β-CD,GLI/γ-CD,GLI/HP-β-CD and GLI/Me-β-CD aggregates were 152.10±28.62(67.2%)nm,118.20±46.43(87.7%)nm,76.77±19.25(13%)nm and 66.14±14.35(13.9%)nm respectively.β-CD andγ-CD had bigger aggregation capability because their-OH groups participated in self-aggregation rather than solvation.Our results were somewhat similar to the previous publication about the same topic since we found that the aggregation size of 2,5-Diphenyloxazole/β-CD was 172.5 nm[26].β-CD andγ-CD had bigger aggregation capability because its-OH groups participated in self-aggregation rather than solvation[60].Theγ-CD had stronger aggregation capability thanβ-CD becauseγ-CD has a high intrinsic solubility.The aggregates of HP-β-CD and Me-β-CD were quite small because the partial substitution of-OH and-CH3groups of CDs prevents the hydrogen-bond formation of the aggregation[59-62].The size measurement results would be compared with the modeling results.The hydrogen bonds is important to the aggregation mechanism.
3.2.2.MD simulation of 10:10 GLI/CD aggregation systems and the comparison of experimental and modeling results
The 10:10 simulation of GLI/CD MD simulation was used to investigate the real state of high-concentration solution.The higher concentration in 10:10 modeling was adjusted by added more drugs and reduce water molecules compared to 1:1 MD simulation.
Fig.6 showed initial structures and final structures of 10:10 systems.GLI/Me-β-CD aggregated three piles,GLI/HPβ-CD aggregated two piles while GLI/γ-CD and GLI/β-CD aggregated only one big pile.The aggregates of GLI and CDs had no regular shape.The final structures contained free drug molecules,“empty”CD molecules,drug/CD inclusion complexes and drug/CD non-inclusion complexes.Our research agreed with the previous theory about the existence of micelle-like structure[20,21,26,32,58,63].
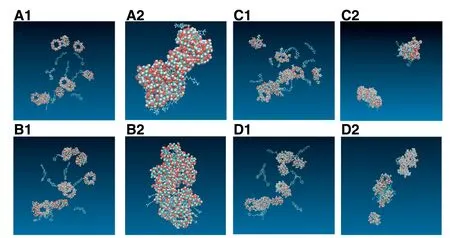
Fig.6-Snapshots initial and final structures of GLI/CD solution.Carbon elements are indicated in blue bonds,hydrogen elements in white,and oxygen elements in red.(A1)initial structure of GLI/β-CD(A2)final structure of GLI/β-CD,(B1)initial structure of GLI/γ-CD,(B2)final structure of GLI/γ-CD.(C1)initial structure of GLI/HP-β-CD,(C2)final structure of GLI/HP-β-CD,(D1)initial structure of GLI/Me-β-CD,(D2)final structure of GLI/Me-β-CD.
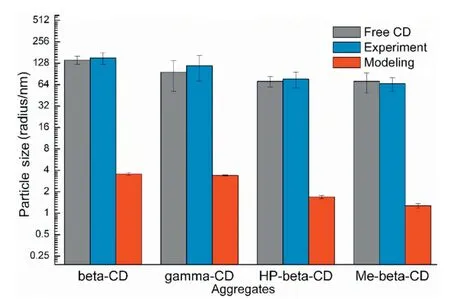
Fig.7-The comparison of aggregation size measured by experimental and modeling method.(Free CD):The aggregation size of free CD measured by experimental method.(Experiment):The aggregation size of drug/CD measured by experimental method.(Modeling):The aggregation size of GLI/CD measured by modeling method.
Fig.7 compared the experimental and simulation results of the aggregation particle size of GLI/CD solution.The particle size of free CD solution was close to those of the GLI/CD solutions,which proved that aggregates were mainly caused by the nature of CDs.The average particle size of GLI/β-CD,GLI/γ-CD,GLI/HP-β-CD and GLI/Me-β-CD simulation systems was:35.60±1.45 ˚A,34.04±0.6 ˚A,16.90±0.89 ˚A and 11.68±0.92 ˚A.Our simulated results also showed thatβ-CD andγ-CD had stronger aggregation capability than HP-β-CD and Me-β-CD,which agreed with the experimental results.However,there were still some differences in the numerical values between experimental and simulation results.The possible reason was that there were millions of molecules in real solutions,which were much larger than our 10:10 simulation systems.Thus,the simulated particles were much smaller than that of the experimental measurement.
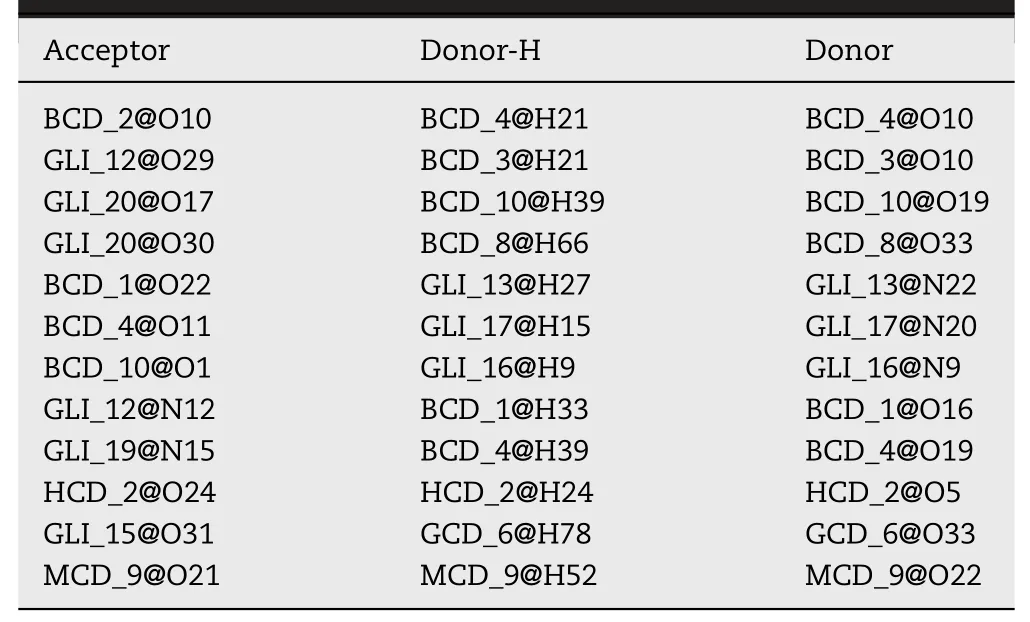
Table 7-Some of the hydrogen bonds information of 10:10 system.(BCD:β-CD,GCD:γ-CD,HCD:HP-β-CD,MCD:Me-β-CD).
The hydrogen bond information in the modeling could explain the mechanisms of aggregation.The aggregation capability of GLI/β-CD and GLI/γ-CD was stronger than those of the GLI/CD-derivates,the mechanism of the conclusion would be investigated by hydrogen bonding analysis.It has been reported that hydrogen bond plays an important role in CD aggregates[64].Table 7 showed acceptor atoms,donor hydrogen atoms and donor atoms of hydrogen bonds in MD simulation.From Table 7,the sulfonyl group,-C=O and pyrazine group of GLI could be hydrogen bond acceptors,while the-NH group of GLI could be hydrogen bond donors.Inβ-CD orγ-CD solution,the-OH groups of CDs were able to form hydrogen bonds with both drugs and CDs.However,the substituted groups of HP-β-CD and Me-β-CD prevented the hydrogen bond formation of aggregates.The simulation results clearly revealed the molecular mechanism of aggregation.
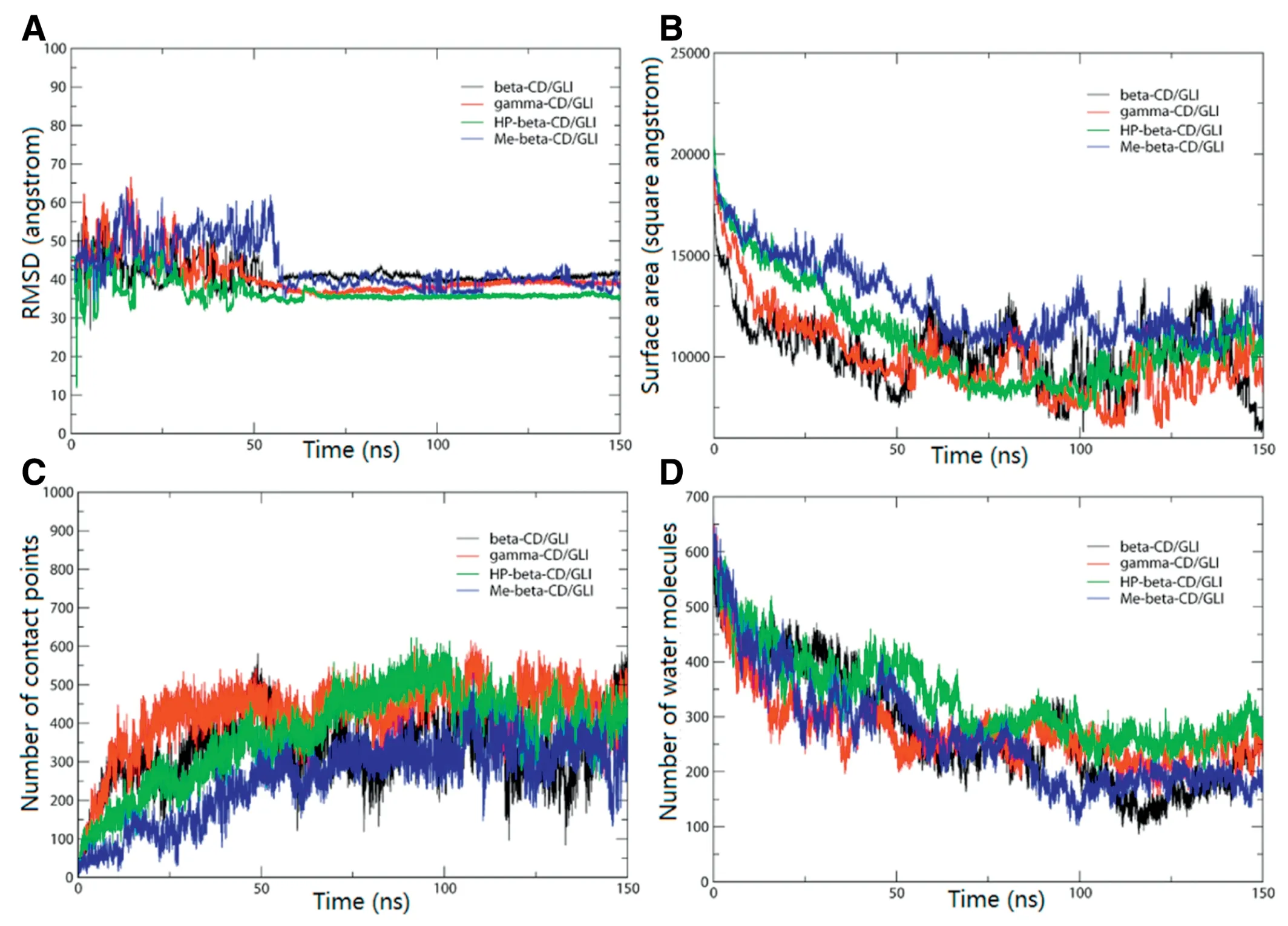
Fig.8-CPPTRAJ analysis results of 10:10 system during 150 ns MD simulation.(A)RMSD curves of GLI/CD 10:10 system.(B)Surface area of GLI/CD complexes.(C)Native contact points between GLI and CDs within 3 ˚A.(D)Water molecules around GLI within 3.6 ˚A.
Fig.8 showed some analysis data of 10:10 GLI/CD system.Fig.8A showed the RMSD plot of mass calculated through 150 ns MD simulation to the original structures.RMSD curves of four systems showed quite stable after 50 ns,which indicated that all systems reached balance after 50 ns.Fig.8B showed the solvent-accessible surface area(SASA)of GLI/CD solution.The solvent-accessible surface area could reflect the degree of aggregation.After about 50 ns,the smaller surface area represented the aggregates.The time dependence of native contact points between GLI and CDs of four systems was shown in Fig.8C.It suggested that the native contact points of GLI and CDs increased in the process of first 50 ns simulation.This behavior reflected a process of aggregation formation.The native contact points changed weakly after 50 ns which confirmed that all systems had been balanced.Fig.8D showed the plot of water molecules around GLI within 3.6 ˚A.The correlation curves decreased significantly which confirmed that more solvent molecules were squeezed away from drugs when the aggregates were formed.GLI was encapsulated by CD molecules while forming aggregates.The decreasing water molecules around GLI showed the physical shielding effect of CDs.All fluctuated curves indicated that the drug/CD complexes aggregates were in the process of dynamic equilibrium after 50 ns.
According to MD simulation of 10:10 GLI/CD aggregation,the experimental and modeling method investigated the aggregation ability of four CDs.The analysis of hydrogen bonds explained the weak aggregation capability of substituent CDs.The other parameters provided us clearer image to the aggregation mechanisms of GLI/CD aggregates.
4.Conclusion
Integrated experimental and molecular modeling methods revealed the molecular inclusion and non-inclusion mechanism of CDs solubility enhancement.Phase solubility studies and simulation results showed that Me-β-CD was the best choice to enhance the solubility of GLI.Moreover,both experimental and modeling results suggested the existence of aggregates in GLI/CD solution.Hydrogen bond plays an important role in drug/CD aggregation.β-CD andγ-CD had strong aggregation capability than HP-β-CD and Me-β-CD.MD simulation provided us a clear image of the molecular mechanism of GLI/CD inclusion complexation and aggregation.
Our integrated experimental and modeling methodology could fit other drugs and CDs.The systems under various conditions(e.g.pH,salt and temperature)could also be mimic in the simulations.Our results will benefit the future formulation development of CD systems.
Conflicts of interest
The authors declare that there is no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgment
This study was supported by the University of Macau Research Grants(MYRG2016-00038-ICMS-QRCM and MYRG2016-00040-ICMS-QRCM).This work was performed in part at the High-Performance Computing Cluster(HPCC)which is supported by Information and Communication Technology Office(ICTO)of the University of Macau.
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Cancer nanotechnology:Enhancing tumor cell response to chemotherapy for hepatocellular carcinoma therapy
- The functions and applications of A7R in anti-angiogenic therapy,imaging and drug delivery systems
- Evaluation of the Mrp2-mediated flavonoid-drug interaction potential of quercetin in rats and in vitro models
- A novel oral prodrug-targeting transporter MCT 1:5-fluorouracil-dicarboxylate monoester conjugates
- Synthesis,characterization and in vivo evaluation of honokiol bisphosphate prodrugs protects against rats’brain ischemia-reperfusion injury
- Improved dissolution and oral absorption by co-grinding active drug probucol and ternary stabilizers mixtures with planetary beads-milling method