GC-MS/MS 法测定桂丁香中β-谷甾醇含量
2020-01-06支荣荣周会芹
支荣荣,周会芹,郑 灏
盐城市食品药品监督检验中心,盐城 224055
桂丁香为樟科植物肉桂Cinnamomum cassia Presl.的干燥带宿萼的未成熟果实,别名肉桂子、桂丁等,入药始见于《百草镜》,《纲目拾遗》引其曰:桂丁“形如吴茱萸,出广西交趾,乃肉桂子也”,所述与今相符。主产广西、广东、福建。具有温里散寒、止痛、止呃的功效[1]。桂丁香含挥发油,油中含桂皮醛(Cinnamaldehyde)、β-谷甾醇(β-sitosterol)、香豆精(coumarin)、胆碱(choline)等[2]。
桂丁香为江苏省地方中药材,收载于《江苏省中药饮片炮制规范》(2002 年版)。本次“标准”修订提出了增加组织显微鉴定、薄层色谱鉴别和HPLC 法测定桂皮醛含量的改进草案。桂丁香中的β-谷甾醇具有抗炎、止咳、抗溃疡、保护胃黏膜、抗癌、抗氧化和降低胆固醇及降血糖的药理作用[3-5]。胡曙晨等[6]建立了β-谷甾醇的薄层色谱鉴别方法,目前还没有报道桂丁香中β-谷甾醇的含量测定方法,为进一步提高质控标准,现以β-谷甾醇含量为指标,建立其含量测定方法。
1 仪器与药品、试剂
1.1 仪器
Agilent7890D 型气相色谱仪,7000D 三重四极杆质谱检测器;BP211D、BT224S 电子天平(德国Sartorius)。
1.2 药品与试剂
桂丁香样品8 批,购自亳州药材市场及盐城市中药饮片有限公司,经原江苏省食品药品监督检验研究院狄恒建主任中药师鉴定确认。
β-谷甾醇对照品(纯度98%,中国食品药品检定研究院);其余试剂为分析纯;水为纯净水。
2 方法与结果
2.1 仪器条件
2.1.1 色谱条件 色谱柱:安捷伦HP-5MS 毛细管柱(30 m×0.32 mm,0.25 μm);进样口温度:320 ℃,检测器温度:345 ℃,程序升温(起始温度160 ℃,以20 ℃·min-1升至290 ℃,保持15 min);分流比:8∶1,进样量:1 μL。
2.1.2 质谱条件[7,8]EI 源;电子轰击能量70 eV;离子源温度300 ℃;四级杆温度150 ℃;质谱接口温度280℃;溶剂延迟时间6min;碰撞气(氮气)流速1.5mL·min-1;猝灭气(氦气)流速2.25 mL·min-1;采用多反应监测模式(MRM),对β-谷甾醇进行一级质谱全扫描〔质荷比(m/z)50~500〕,选择响应较强的质荷比(m/z)为329.0 的碎片作为母离子,再对该母离子进行子离子扫描〔质荷比(m/z)50~329〕,确定定性离子对(m/z)329.0/95.2 和定量离子对329.0/189.2。
质谱扫描图见图1~2,对照品溶液及供试品溶液质谱图见图3~4。
2.2 溶液的制备
2.2.1 对照品溶液 精密称取β-谷甾醇对照品9.94 mg,置50 mL 量瓶中,加二氯甲烷溶解、稀释至刻度,混匀,作为对照品贮备液(0.198 8 mg·mL-1)。精密量取贮备液1 mL 置10 mL 量瓶中,加二氯甲烷稀释至刻度,混匀,作为对照品溶液(19.88 μg·mL-1)。
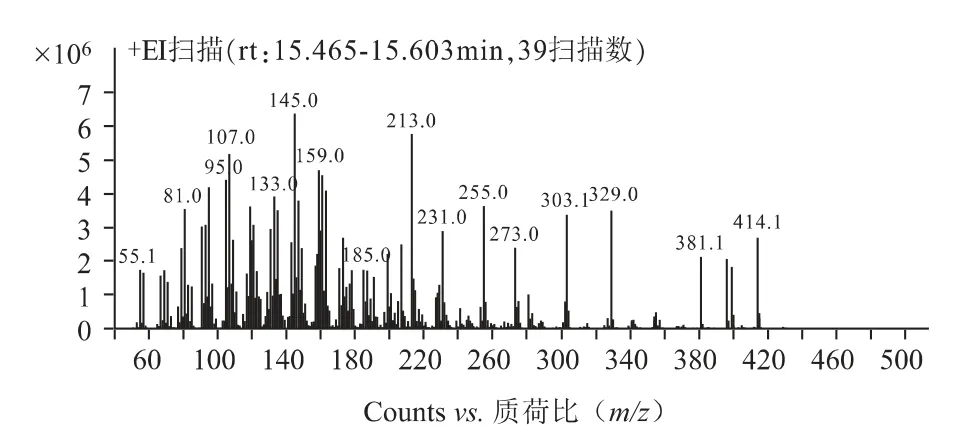
图1 β-谷甾醇一级全扫描质谱图
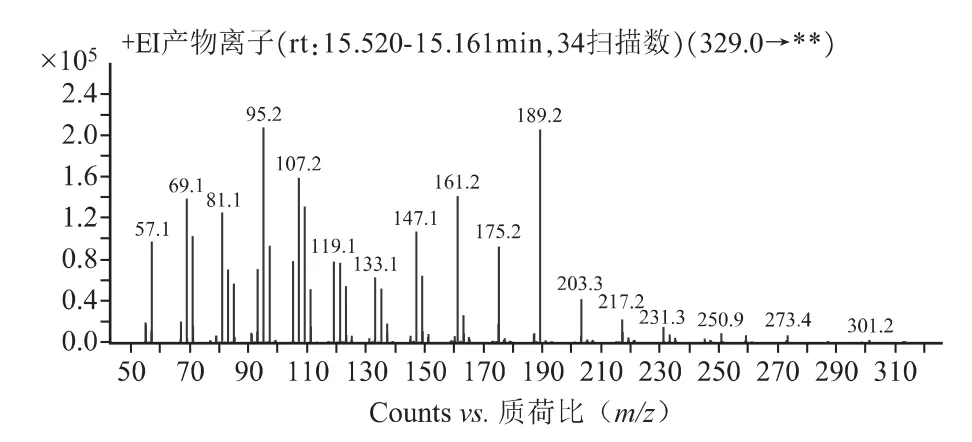
图2 母离子扫描质谱图
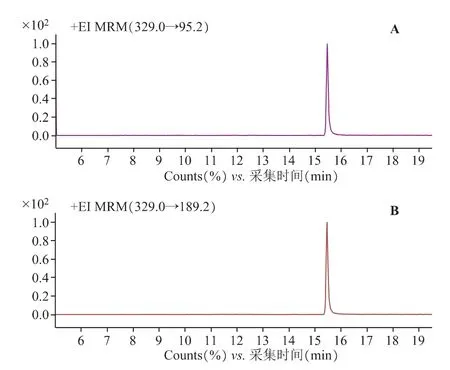
图3 β-谷甾醇对照品质谱图
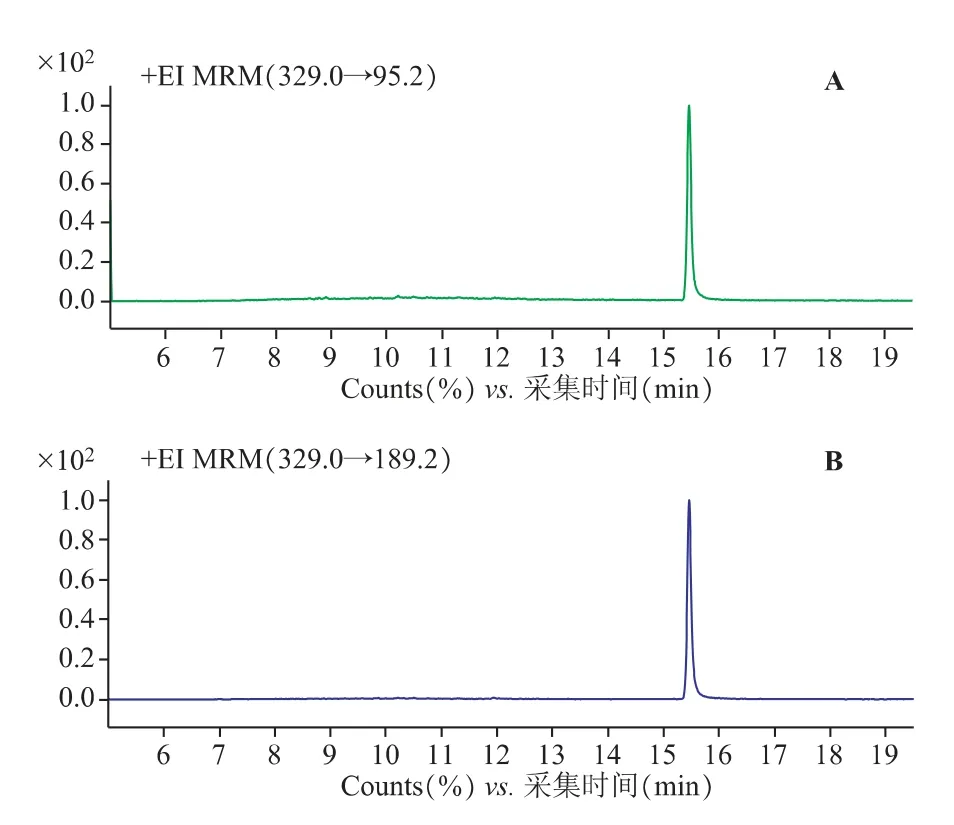
图4 桂丁香供试品质谱图
2.2.2 供试品溶液 取本品粉末(过3 号筛)2.5 g,精密称定,加入二氯甲烷适量,回流至提取液近无色(约3 h),收集提取液,回收溶剂至干,固体物加二氯甲烷适量溶解,定容至10 mL 量瓶中,精密量取5 mL,加在中性氧化铝柱上,用二氯甲烷50 mL洗脱,收集洗脱液,用0.45 μm 微孔滤膜滤过,取续滤液,即得。
2.3 线性范围考察
分别精密吸取对照品贮备液0.25、0.5、0.75、1、1.5、2、2.5 mL,置10 mL 量瓶中,加二氯甲烷至刻度,混匀。精密吸取上述溶液1 μL,注入气相色谱仪,测得峰面积。以β-谷甾醇浓度为横坐标,以峰面积为纵坐标,绘制标准曲线,计算得线性回归方程:Y=1.2842×104X-9.5718×104,(r=0.999 5)。
结果表明,β-谷甾醇在4.97~49.7 ng 范围内与峰面积呈良好的线性关系。
2.4 进样精密度试验
精密吸取对照品溶液(19.88 μg·mL-1),按“2.1”仪器条件测定,重复进样6 次,平均峰面积(n=6)为25 743,RSD 为1.2%,表明精密度良好。
2.5 稳定性考察
按“2.2.2”项方法制备供试溶液,按“2.1”仪器条件测定,分别于0、4、8、12、16、24 h 进样,平均峰面积(n=6)为22 806,RSD 为1.1%,结果表明供试品溶液在24 h 内稳定。
2.6 重复性试验
精密称取同一供试品,按“2.2.2”项方法制备供试品溶液6 份,按“2.1”仪器条件测定,β-谷甾醇的平均含量(n=6)为0.077%,RSD 为1.6%,结果表明重复性良好。
2.7 加样回收率试验
精密称取已知含量的样品(含量0.077%,以干燥品计)1.25 g,共6 份,分别精密加入对照品贮备液(0.198 8 mg·mL-1)5 mL(含β-谷甾醇0.994 0 mg),按“2.2.2”项方法制备供试品溶液,按“2.1”仪器条件测定,计算回收率,结果见表1。
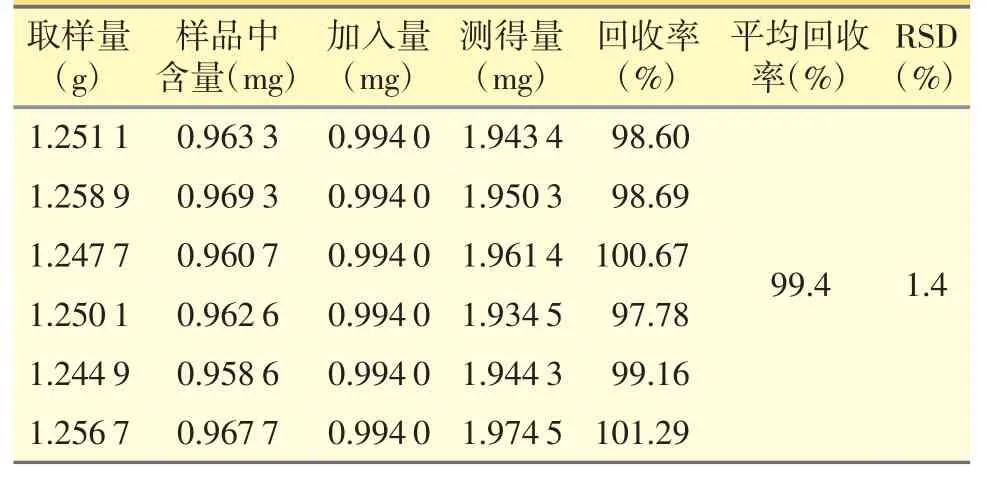
表1 加样回收试验结果(n=6)
2.8 样品测定
取8 个不同批次桂丁香样品,按照“2.2.2”项方法制备供试品溶液,按“2.1”仪器条件测定,以干燥品计算,各批次含量分别为0.038%、0.050%、0.077%、0.078%、0.087%、0.062%、0.081%、0.071%。
3 讨论
3.1 测定方法的选择
β-谷甾醇无紫外最大吸收,蒸发光散射法、火焰离子化气相色谱法(FID)的灵敏度较低,因此,采用气相色谱-串联质谱法(GC-MS/MS)测定β-谷甾醇含量,以多反应离子监测模式(MRM)采集信号,具有专属性强、检测灵敏度高、重复性好等特点。
3.2 色谱条件的确定
在使用GC-MS/MS 测定β-谷甾醇之前,首先采用FID 检测器对β-谷甾醇进行色谱条件的优化,发现柱温对β-谷甾醇的峰形及分离度有较大影响。尝试采用270℃柱温[7,8]时β-谷甾醇峰形较好,但是有一杂质峰与待测成分分离度不能达到要求,当温度提高到290 ℃时,该杂质与待测成分完全分离(安捷伦HP-5 毛细管柱可以耐受最高350 ℃)。
进样口温度按照《中国药典》要求高于柱温30~50 ℃,因此选择了320 ℃,检测器温度一般高于进样口温度,选择了345 ℃,程序升温(起始温度160 ℃,以20 ℃·min-1升至290 ℃,保持15 min),以此可先分离出杂质碎片。分流比的选择考察了2∶1、4∶1、8∶1、10∶1、20∶1;分流比过大,响应值低,分流比过小,有拖尾现象,因此选择8∶1。
3.3 供试品溶液的制备
因供试品杂质较多,β-谷甾醇的响应值较低,采用二氯甲烷索氏提取,提取液浓缩至10 mL 的方法,减少稀释倍数,提高待测成分的浓度,用FID 检测器,致β-谷甾醇响应值有所提高,可达到定量限要求。供试品浓度太大,杂质太多,对色谱柱与检测器的耐受性有影响,因此选择了灵敏度更高的GCMS/MS 进行测定。对供试品作了进一步稀释,并增加了柱层析的步骤,有效减少了杂质的影响。
