基于QTL定位和全基因组关联分析筛选甘蓝型油菜株高和一次有效分枝高度的候选基因
2020-01-02陈志友荐红举曲存民李加纳
霍 强 杨 鸿 陈志友 荐红举 曲存民 卢 坤 李加纳,*
基于QTL定位和全基因组关联分析筛选甘蓝型油菜株高和一次有效分枝高度的候选基因
霍 强1,2,**杨 鸿1,2,**陈志友1,2荐红举1,2曲存民1,2卢 坤1,2李加纳1,2,*
1西南大学农学与生物科技学院/ 油菜工程研究中心, 重庆 400715;2西南大学现代农业科学研究院, 重庆 400715
株高和一次有效分枝高度是与甘蓝型油菜结荚层厚度、收获指数紧密关联的重要农艺性状, 有关株高的数量性状位点(quantitative trait locus, QTL)和全基因组关联分析(genome-wide association study, GWAS)已有很多报道, 但对一次有效分枝高度的QTL和GWAS定位以及候选基因筛选的研究报道较少。本研究利用已构建的高密度遗传连锁图对2016和2017年2个环境的186个株系组成的重组自交系群体株高和一次有效分枝高度及其最佳线性无偏预测(best linear unbiased prediction, BLUP)值进行QTL定位共检测到8个株高的QTL, 分别位于A03、A04和A09染色体, 单个QTL解释4.60%~13.29%的表型变异, 其中位于A04染色体上的QTL (q-2017PH-A04-2和q-BLUP-PH-A04-2)在2017年和BLUP中均被检测到; 检测到9个一次有效分枝高度QTL, 分别位于A01、A02、A05、A09、C01和C05染色体上, 单个QTL解释5.12%~19.10%的表型变异, 其中q-2017BH-A09-1、q-BLUP-BH-A09-2和q-BLUP-BH-A09-3有重叠区段。同时, 利用课题组前期完成的588份重测序自然群体进行全基因组关联分析, 2年共检测到与株高显著关联的50个SNP位点和与一次有效分枝高度显著关联的12个SNP位点; 根据SNP的物理位置, 筛选出参与细胞增殖、细胞扩增、细胞周期和细胞壁活动的13个株高候选基因, 以及参与赤霉素、亚精胺等合成代谢途径、核糖体组成和在光合、萌发等过程中有一定作用的一次分枝高度的11个候选基因, 并利用荧光定量PCR技术验证候选基因在极端材料中的表达情况。本研究结果将为油菜株型改良及后续基因的功能研究提供理论依据。
甘蓝型油菜; 株高; 一次有效分枝高度; 基因定位; 候选基因筛选
甘蓝型油菜是四大油料作物之一, 且在饲料、绿肥、蔬菜、能源、旅游、蜜源等方面都有重要价值。作物的株型结构在作物从幼苗到成熟的生长期间以及作物对环境条件的响应中发生变化, 这种可塑性是由作物发育的灵活变化带来的[1], 同时作物株型的变化对作物的适应性和产量具有显著影响[2-3]。
株高是作物株型最突出的决定因素, 在作物中常被驯化或选择。其中矮秆基因已广泛应用于提高谷类作物的抗倒伏能力和收获指数[4]。第一次“绿色革命”, 将半矮化基因引入水稻()和矮化基因引入小麦()来降低作物株高[5], 使2种作物增加其种植密度, 通过提高收获指数而大幅增产。拟南芥中,()、()、()、、是5个DELLA蛋白基因, 突变体和在DELLA蛋白结构域的序列缺失导致植株严重矮化[6-7]。和基因突变植株也表现出矮化现象[8]。作物的分枝同样是影响株型的重要因素。玉米的基因可抑制侧生器官生长[9], 在小麦中过表达此基因可抑制小麦分蘖发育[10]。番茄()基因[11]、拟南芥基因[12]和水稻()基因[13]是同源基因, 以及番茄中()基因[14], 它们的突变可显著减少营养分枝和生殖分枝的数量; 此外, 拟南芥()、豌豆()、矮牵牛()等基因也对分枝生长发育有着重要作用[15]。截至目前, 株高候选基因在模式植物中虽已经得到大量的克隆和鉴定, 但在甘蓝型油菜中仅有几例报道[16], 且一次有效分枝高度基因在甘蓝型油菜中尚无定位和克隆的报道。
利用QTL和GWAS相结合是定位数量性状候选基因的有效方法[17]。王嘉等[18]利用RIL (recombinant inbred lines)群体检测到分布于A01、A06、A07、A08、A10和C06染色体上的11个株高相关QTL, 单个QTL可解释的表型变异为5.00%~15.26%; 并检测到7个一次有效分枝高度相关QTL, 分别位于A06、C05和C06染色体上, 单个QTL解释5.04%~12.99%的表型变异。Zhao等[19]利用1个DH (doubled haploid)群体, 检测到18个位于A02、A03、A07、A10、C01、C03、C04、C06和C09染色体上的株高相关QTL和27个位于A01、A02、A03、A06、A07、A09、A10、C03、C05、C06和C09染色体上的一次有效分枝高度相关QTL。Cai等[2]利用DH群体, 检测20个株高相关QTL和16个一次有效分枝高度相关QTL。Luo等[20]对(a doubled-haploid population)进行基因分型, 并对该群体之前的表型重新分析, 检测到80个株高相关QTL, 分布在除C01外的各个染色体上; 同时检测到35个与一次有效分枝高度相关QTL, 分布在A01、A02、A03、A05、A06、A09、A10、C05、C06、C07、C08、C09上, QTL的贡献率为3.19%~22.91%。贺亚军等[21]利用1个DH群体和1个永久性F2群体, 检测到9个株高相关QTL, 分布于A02、A09、C01、C02和C06连锁群上; 同时检测到11个一次有效分枝高度相关QTL, 分布于A01、A03、A09、C01和C03连锁群上, 单个QTL可解释4.01%~16.54%的表型变异。Shen等[22]利用DH群体检测到5个株高相关QTL和5个一次有效分枝高度相关QTL, 位于A02和A07染色体上。但以上报道均以单个方法筛选候选基因, 而使用QTL定位和全基因组关联分析(GWAS)筛选和鉴定候选基因具有重要意义。
虽然株高和一次有效分枝高度的相关基因挖掘已经有较多的报道[23-25], 但是本文利用不同的作图群体和具有物理位置的SNP标记, 筛选QTL和显著关联的SNP, 并利用其物理位置信息筛选候选基因, 结合qRT-PCR结果进一步验证筛选的候选基因, 这将为基因功能分析奠定基础。
1 材料与方法
1.1 试验材料
用于QTL定位的群体是以GH06为母本、中油821 (ZY821)为父本杂交获得F1, 自交的F2代通过“一粒传法”连续自交10代以上构建形成含186个株系的高世代重组自交系群体。该群体材料于2015—2016年度、2016—2017年度连续2年种植于重庆市北碚区歇马镇油菜基地(29ºN, 106ºE, 海拔238.6 m), 以育苗移栽方式, 随机区组设计, 2个重复, 每小区种植3行, 每行15株, 行距40 cm, 株距20 cm。按照常规生产方式进行田间管理。
用于全基因组关联分析的自然群体是由全球油菜主要生产地征集的588份油菜品系组成[26], 其中大部分来自国内重庆、湖北、湖南、江苏、陕西等地, 部分来自加拿大、德国、瑞典、丹麦、澳大利亚等国家。该群体所有种植及田间管理同重组自交系群体。
1.2 性状考察
成熟期, 在每个小区中部选择5株长势一致的油菜植株统计调查表型性状, 分别测定其株高(plant height, PH)、一次有效分枝高度(the first branch height, BH)、结荚厚度(thickness of pod canopy, TPC)、经济产量(economic yield, EY)和收获指数(harvest index, HI)。株高指从子叶节到整株油菜主茎最高点的长度, 单位为cm; 一次有效分枝高度是指子叶节与主茎上最下部第一个有效分枝着生点之间的距离, 单位为cm; 结荚厚度是指整株油菜最高一个有效角果着生点与最低一个有效角果着生点的高度差, 单位为cm; 经济产量是指5株植株所有自然风干种子总重量的平均值, 以“g”为单位; 收获指数是指5株油菜单株经济产量之和与其生物产量之和的百分比。
1.3 表型数据分析
利用Microsoft Excel 2007对高世代重组自交系群体和自然群体的表型数据进行整理和简单的统计分析; 利用IBM SPSS Statistics 19统计分析软件进行描述性统计分析并制作正态分布图、进行相关分析; 变异系数: CV =/, 其中为标准差,为平均值; 广义遗传力:2=2G/(2G+2GE/+2e/), 其中2G表示基因型方差,2GE表示基因型与环境互作方差,2e为误差,代表环境数目,代表每个环境的重复数; 利用R软件对两年两群体的表型数据进行最佳线性无偏预测(best linear unbiased prediction, BLUP)。
1.4 遗传图谱定位方法
取每个株系的5个幼嫩叶片混合, 提取DNA用于SNP标记分析。按照Illumina公司的Infinium HD Assay Ultra的说明进行DNA样品的预处理、与芯片杂交、洗脱、单碱基延伸、染色及包埋, 芯片准备好后利用Illumina HiSCAN扫描, 并利用GenomeStudio genotyping software v2011对扫描结果进行分析获得每个株系的基因型, SNP遗传图谱包括8575个SNP标记, 1201个bin, 覆盖甘蓝型油菜基因组6140.2 cM。采用WinQTL Cart2.5软件[27]的复合区间作图程序进行株高和一次有效分枝高度的QTL定位和效应检测。进行CIM分析时, 设置1 cM的步长(walking speed), 1000次回归, 显著水平0.01。遵照McCouch等[28]的方法命名QTL, 斜写的小写字母“q”加上性状的名字, 后面跟染色体, 最后一个数字表示QTL的序号。如q-2016PH-A01-1表示2016年株高在A01染色体上的第1个QTL。
1.5 全基因组关联分析的方法
1.5.1 基因型数据测定与分析 对588份甘蓝型油菜进行重测序, 数据包含134.3亿个150 bp的双末端片段, 过滤后大约有530万个SNP。通过基因型分析, 最终获得385,692个可利用的SNP标记数据[26], 这些标记数据的最小基因型频率(minor allele frequency, MAF)大于0.05, 利用这些覆盖甘蓝型油菜全基因组的高密度标记, 对甘蓝型油菜自然群体进行群体结构(Q)、亲缘关系(K)和连锁不平衡(LD)分析, 并用这些标记结合重庆2年的株高和一次有效分枝高度的表型数据进行全基因组关联分析。
1.5.2 群体结构、亲缘关系分析与连锁不平衡分析
利用Structure 2.3.4软件把每份甘蓝型油菜材料归类到特定的2个亚群里, 以此确定总群体的群体结构。运用TASSEL 5.0软件对自然群体中任意的2份材料进行亲缘关系的评估, 计算两特定材料间的遗传相似度与任意材料间的遗传相似度的相对值, 用0替代系数小于0的相对值。本群体中约2/3的材料间亲缘关系值小于0.05, 群体材料间亲缘关系越弱, 对进一步关联分析的影响越小[29]。运用TASSEL 5.0软件对群体内连锁不平衡(LD)进行分析计算。利用染色体上非等位基因位点间的2值来估算甘蓝型油菜染色体中A和C染色体组LD的衰减, A和C染色体组的LD随着物理距离的增加而下降, 但A和C染色体组的衰减程度不同, 在同一物理位置A染色体组比C染色体组衰减快[26]。
1.5.3 全基因组关联分析 采用naive、Q、PCA、K、Q+K和PCA+K六种统计模型来评估群体结构、亲缘关系的影响进行表型和SNP关联分析(Q: 群体结构; PCA: 主成分; K: 亲缘关系)。运用TASSEL 5.0软件进行这6种模型的关联分析, 其中使用一般线性模型(GLM)分析naive、Q和PCA模型, 使用混合线性模型(MLM)分析K、Q+K和PCA+K模型。利用SAS软件对这6种模型的运算结果, 对其-lg ()的观测值和-lg ()期望值绘制Quantile-quantile散点图, 通过QQ图比较后确定最佳模型, 在最佳模型下进行株高和一次有效分枝高度性状的全基因组关联分析, 利用R软件作Manhattan图。本试验使用的SNP数据为385,692个,值小于阈值(1/385,692 = 2.593E-06)的位点为显著关联位点。对于GWAS结果, 定位区间为显著关联SNP位点左右延伸500 kb[30]。
1.6 候选基因的筛选
根据甘蓝型油菜“Darmor-bzh”参考基因组[31], 提取株高和一次有效分枝高度性状定位的QTL置信区间内的基因; 根据分析得到的株高和一次有效分枝高度关联SNP标记, 提取各位点500 kb内的基因; 将需要功能分析的基因序列与拟南芥所有基因序列进行BLASTN比对, E-value阈值为1E-10, 并以同源性最高的拟南芥基因作为待分析基因的功能注释来筛选与株高和一次有效分枝高度相关的候选基因。
1.7 候选基因的验证
对株高和一次有效分枝高度极端材料在蕾薹期取3个生物学重复的茎尖并提取RNA。根据前期所取材料的茎尖提取的RNA, 使用TaKaRa公司的PrimeScript RT reagent Kit with gDNA Eraser反转录合成cDNA, 将得到的cDNA产物稀释10倍后作为模板, 用于qRT-PCR验证。以油菜内参基因作为对照, 使用TaKaRa公司的SYBR Premix ExII试剂盒进行qRT-PCR验证候选基因的相对表达量。设置每个样品3次技术重复。在Bio-Rad定量PCR仪上进行qRT-PCR扩增。在网站http://biodb. swu.edu.cn/qprimerdb上查找设计候选基因的qRT-PCR引物(表1), 由上海生工生物工程有限公司合成。
2 结果与分析
2.1 重组自交系群体与自然群体的表型变异
重组自交系群体中, 亲本GH06和ZY821的株高分别为198.1 cm和186.6 cm, 一次有效分枝高度分别为90.7 cm和67.8 cm,检验株高在亲本间未达到显著差异(>0.05), 但一次有效分枝高度达到显著差异(<0.01)。株高在2016年和2017年的变异系数分别为5.24%和5.75%, 一次有效分枝高度的变异系数为18.58%和17.73%; 自然群体中, 株高在2016年和2017年的变异系数分别为8.57%和10.16%, 一次有效分枝高度的变异系数分别为26.65%和27.86% (表2)。2年间两性状的变异系数较为稳定, 株高的变异系数相对于一次有效分枝高度较小。株高在重组自交系和自然群体中的广义遗传力分别为75.45%和87.42%, 一次有效分枝高度分别为68.77%和82.21%; 在同一群体中, 株高较一次有效分枝高度的遗传力高。自然群体中, 测量株高和一次有效分枝高度极端材料YC4、YC15、YC11的表型数据, YC4和YC15株高分别为213.9 cm和185.7 cm; YC11和YC4的一次有效分枝高度分别为100.8 cm和70.3 cm, 2年极端材料的株高及一次有效分枝高度都达到显著差异(<0.01)。
重组自交系群体和自然群体2年株高和一次有效分枝高度均表现出连续正态分布(图1), 说明这2个性状都符合由多基因控制的数量性状特点, 满足QTL作图的基本要求, 适合全基因组关联分析。
2.2 重组自交系群体QTL定位分析
利用WinQTL2.5对株高和一次有效分枝高度共检测到19个QTL, 分布在8条染色体上, 其中在A09染色体上检测到的QTL数量最多(图2)。位于A01和A09上的QTL加性效应为负值, 表明这些QTL的增效基因来自父本, 其余的QTL则来自母本(表3)。
对于株高, 2016年检测到2个QTL, 2017年检测到6个QTL, 基于2年数据的BLUP值检测到3个QTL, 其中(A04: 13,373,664–13,531,885)和(A04: 13,373,664–13,531,885)重合,(22,590,345–23,259,990)(23,517,031–23,521,212)(23,603,951– 23,642,701)有交叉重叠区段, 因此共检测到8个QTL。这些QTL主要分布于A03、A04、A09染色体, LOD值介于2.60~5.20, 表型变异贡献率为4.60%~13.29%。
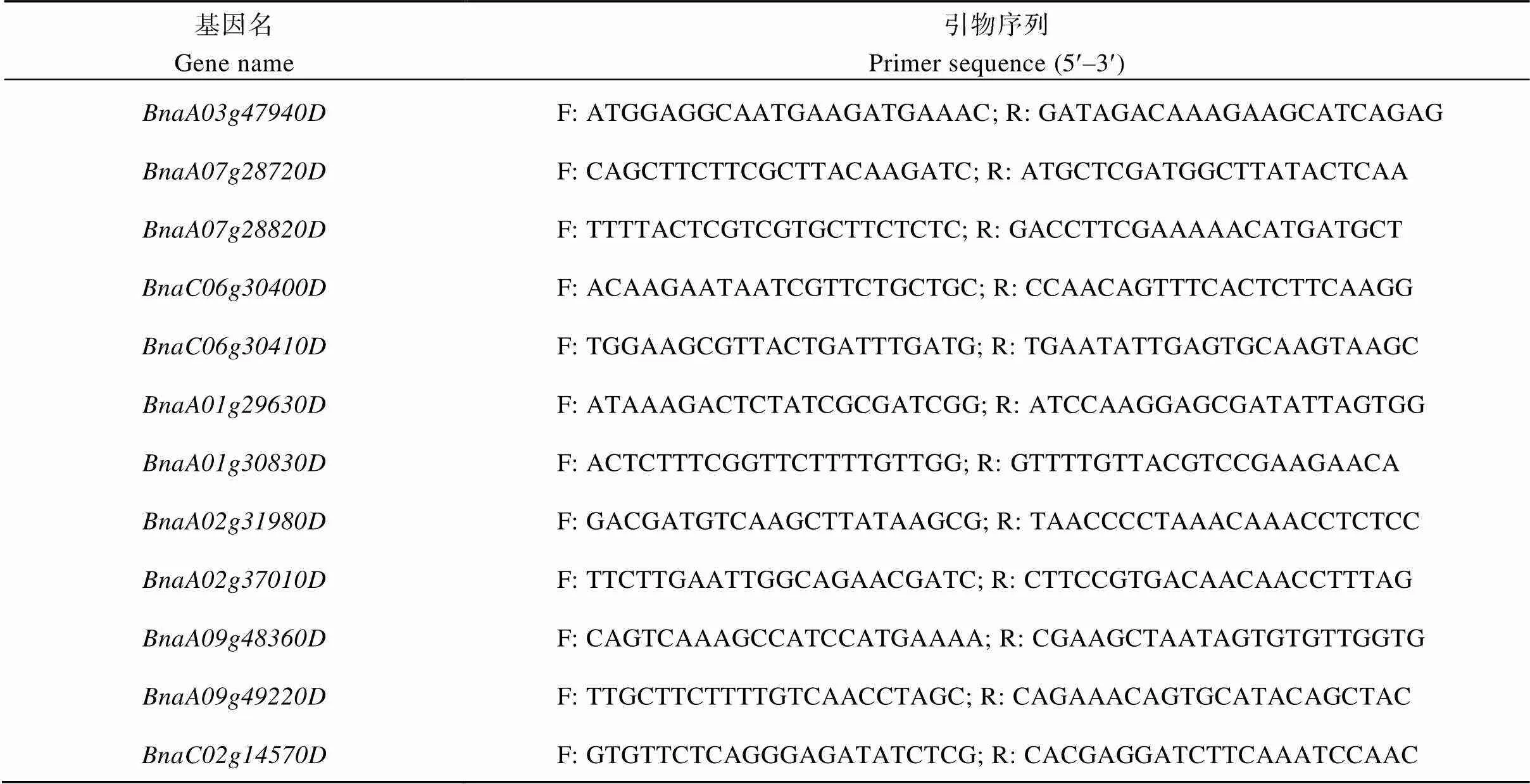
表1 候选基因引物及其扩增产物

表2 两群体株高和一次有效分枝高度的表型数据
RIL: 重组自交系; N: 自然群体。
PH: plant height; BH: the first branch height; SD: standard deviation; CV: coefficient of variation;2: broad sense heritability; RIL: recombinant inbred lines; N: natural population;♂: male parent;♀: female parent.

图1 两群体株高和一次有效分枝高度的频次分布
PH: 株高; BH: 一次有效分枝高度; RIL: 重组自交系; N: 自然群体。
PH: plant height; BH: the first branch height; RIL: recombinant inbred lines; N: natural population.

图2 株高和一次有效分枝高度QTL在SNP连锁群上分布情况
对于一次有效分枝高度, 在2016年和2017年分别检测到2个和4个QTL, 基于2年数据的BLUP值检测到5个QTL, 其中(32,375,997− 32,618,214)(32,435,925−32,608,817)(32,514,368−32,619,386)有交叉重叠的区段, 因此共检测到9个QTL。这些QTL主要分布于A01、A02、A05、A09、C01和C05染色体, LOD值介于2.76~6.52, 表型变异贡献率为5.12%~19.10%。
2.3 自然群体的全基因组关联分析
对株高和一次有效分枝高度关联分析6种模型下的结果绘制Quantile-Quantile散点图(QQ plot), 将各模型下的曲线与期望曲线拟合度最高的模型作为最佳模型。由图3可知, 各性状的最佳模型如下, 株高(PH)都为P+K模型, 一次有效分枝高度(BH)除2016年为K模型外, 其余也为P+K模型。在最佳模型下, 利用385,692个SNP, 以值小于阈值(1/385,692 = 2.593E-06)确定显著关联SNP位点(附表1), 并绘制Manhattan图(图3)。
对于株高, 2016年在A03染色体上检测到1个SNP; 2017年检测到49个SNP, 分别位于A02 (1个)、A07 (45个)和C06 (3个)染色体; 2年BLUP也检测到67个SNP, 分别位于A02 (2个)、A07 (59个)、C02 (1个)和C06 (5个)染色体。在这些位点中, 有42个SNP在2017年和基于BIUP都被检测到, 因此对于株高共检测到75个SNP, 它们的贡献率为6.01%~20.05%, 其中2016年A03染色体上的S3_24415510位点贡献率最大。

表3 在两年中检测到的株高和一次有效分枝高度QTL
BLUP: 最佳线性无偏预测。
PH: plant height; BH: the first branch height;BLUP: best linear unbiased prediction.
对于一次有效分枝高度, 2016年在C03(2个)、C04(1个)和C09(1个)染色体上检测到4个SNP; 2017年检测到8个SNP, 分别位于A02(6个)、C02(1个)和C05(1个)染色体; 基于2年BLUP检测到7个SNP, 分别位于A02(5个)、C02(1个)和C05(1个)染色体。其中有5个SNP在2017年和BIUP中都被检测到, 因此共检测到14个一次有效分枝高度SNP。这些SNP的贡献率为6.23%~18.36%, 其中2016年C04染色体上的S14_36535069位点贡献率最大。株高和一次有效分枝高度在GWAS和遗传连锁定位结果中均没有重合位点。
2.4 候选基因筛选
本文将所得的QTL置信区间内的基因与关联分析SNP位点上下游500 kb区间内的基因比对[30], 初步筛选出24个与油菜株高和一次有效分枝高度相关的基因。并将其基因序列与拟南芥基因序列进行BLAST比对, 结合前人已报道的拟南芥同源基因功能, 筛选可能在本研究中发挥作用的候选基因(表4)。这些基因在拟南芥同源基因中参与植物生长发育, 影响细胞增殖、细胞扩增, 调节细胞周期、细胞壁形成, 参与赤霉素、亚精胺等合成代谢途径、核糖体组成等。


表4 株高和一次有效分枝高度候选基因
PH: plant height; BH: the first branch height.
2.5 候选基因的验证
挑选了其中5个株高候选基因进行验证(图4), 其中基因的相对表达量在株高较高的材料(YC4)中较高, 在株高较低的材料(YC15)中较低;基因的相对表达量在株高较低的材料中较高, 在株高较高的材料中较低。
从一次有效分枝高度的候选基因中, 挑选了7个进行验证(图5), 其中基因的相对表达量在一次有效分枝高度较高的材料(YC11)中较高, 在一次有效分枝高度较低的材料(YC4)中较低;基因的相对表达量在一次有效分枝高度较低的材料中较高, 在一次有效分枝高度较高的材料中较低。

图4 株高候选基因在茎尖中的相对表达量
误差线表示3次生物学重复的标准差。*和**分别表示基因在不同材料茎尖中的表达水平有显著(< 0.05)和极显著(< 0.01)差异。
The error bar shows the standard deviation of three biological replicates. * and ** indicate significantly different expression in stem apex to the control at< 0.05 and< 0.01, respectively.
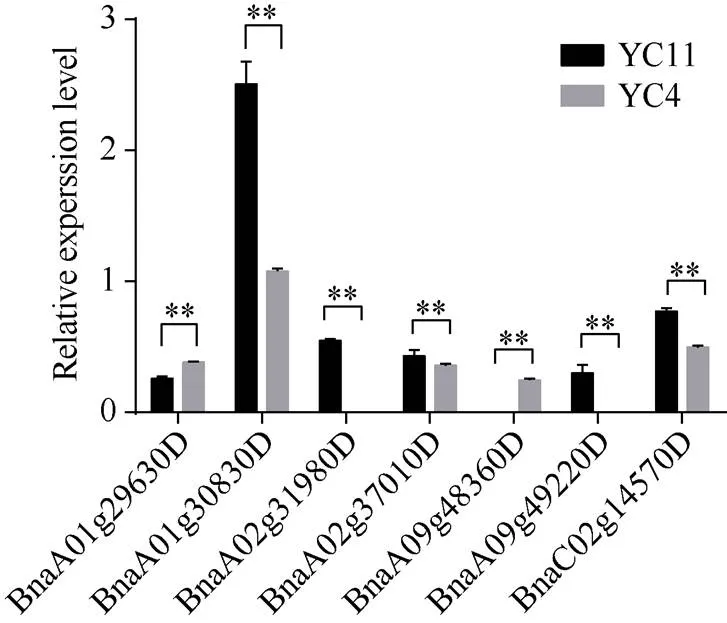
图5 一次有效分枝高度候选基因在茎尖中的相对表达量
误差线表示3次生物学重复的标准差。*和**分别表示基因在不同材料茎尖中的表达水平有显著(< 0.05)和极显著(< 0.01)差异。
The error bar shows the standard deviation of three biological replicates. * and ** indicate significantly different expression in stem apex to the control at< 0.05 and< 0.01, respectively.
3 讨论
株高和一次有效分枝高度是甘蓝型油菜的2个重要株型性状。株高是作物根颈部到顶部之间的距离, 也是影响作物倒伏的重要的因素。一次有效分枝高度是油菜下部第一个有效分枝生长着生点与子叶节之间的距离, 而分枝则作为角果生长的载体。适宜的株高和一次有效分枝高度, 有利于增强植株的抗倒伏性和提高机械化利用率, 进而提高产量并促进油菜产业发展。结荚厚度包含了所有产量构成因素, 经济产量和收获指数又是衡量产量的2个重要指标。
在试验过程中, 对株高和一次有效分枝高度的2016年表型值、2017年表型值和2年的BLUP值都进行了QTL定位和GWAS分析。BLUP能较为有效地利用亲缘表型信息、考虑不同世代的遗传差异并矫正环境效应, 但由于实际中数据的不完整、模型的真实度及其方差组分等的真值未知等问题, 通过BLUP得到的校正值并不一定是最佳的无偏预测, 因此本试验同时将3个值都进行了分析。两性状在2016年和2017年都未检测到重复位点, 利用BLUP值检测到的位点中, 发现与2017年有重复或交叉重叠, 没有与2016年重叠。一方面可能是性状本身受环境的影响较大, 另一方面可能是2016年材料缺失较为严重。
在重组自交系群体中检测到的10个株高性状QTL分别位于A03、A04和A09染色体上, 其中在2016年、2017年和BLIP值中检测到的位于A09染色体上的QTL加性效应均为负值, 说明A09染色体上的QTL增效作用均来自父本的等位基因位点, 且A09上的QTL数量最多, 各位点的贡献率相对较大; 另外, 在2017年和基于BLUP值检测中, A04染色体上有一个重复QTL (q-BLUP-PH-A04-2)。本研究中的q-2016PH-A03-1、q-2017PH-A09-1、q-2017PH- A09-2、q-2017PH-A09-3所在的区段在其他研究中也被重复检测到[21,32-33]。在重组自交系群体中检测到9个一次有效分枝高度性状QTL, 分别位于A01、A02、A05、A09、C01和C05染色体, 其中q-BLUP- BH-C05-1所在区段与王嘉等[18]研究中被检测到的区段有重叠区域。
在自然群体中, 株高性状检测到的A02染色体上的S2_4664084位点和A07染色体上的S7_20033861、S7_20050137等位点与Sun等[24]研究中的位点相近, 最近距离只有7.5 kb; A02染色体上有3个重复SNP, C02和C05染色体上各有1个重复SNP; 一次有效分枝高度性状检测到的A02染色体上的S2_6130942位点与Zheng等[23]研究中的位点相近。
在株高候选基因的拟南芥同源基因中, 检测到的与细胞增殖、细胞扩增、细胞周期和细胞壁相关的基因有。编码的阿拉伯半乳聚糖蛋白是参与植物生长发育的细胞外蛋白多糖[34]。与植物细胞壁形成和重建相关, 在拟南芥的茎生长速率最快阶段,表达增加[35], 由此推测其可能在控制油菜株高性状过程中起关键作用。另外,是一个IAA特异调控的基因, 它在一定浓度IAA处理后表现下调[36-38]; 在一个细胞大小和数量严重减少的矮小拟南芥突变体中,下调[39]。是内切-1,4-β-葡聚糖酶(EGase)基因, 参与细胞伸长, 且与细胞壁的重组和生长有关[40], 由此推测在油菜主茎伸长过程中细胞伸长发挥作用。CEL1蛋白存在于正在发育伸长的幼嫩组织细胞壁中, 特别是在增厚的细胞壁中。CEL1在细胞扩增及增强内切-1,4-β-葡聚糖酶活性与纤维素生物合成方面具有重要作用[41]。编码一个假定的丝氨酸/苏氨酸激酶, 类似Hippo/STE20激酶, SIK1与MOB1相互作用, 调控拟南芥细胞生长和增值, 在突变体中, 细胞数量、细胞大小呈倍性水平降低[42]。编码与CDC2A发生物理作用的d型细胞周期蛋白在种子萌发早期有关键作用并影响细胞分裂。受AUX1和SNLs调控, 在生长中发挥重要作用[43]; 其中在细胞分裂素受体突变体()的全基因组分析中发现下调表达[44], 因此该基因可能通过调控植物激素转导途径来控制油菜株高。
在一次有效分枝高度候选基因的拟南芥同源基因中,编码一种新的亚精胺合成酶, 它是亚精胺合酶和的旁系同源物。和转录水平在根系中高于其他器官, 但在茎节间和胚芽以及根系中的表达都高; 另外发现可能参与ABA介导的胁迫反应[45]。参与了拟南芥GA的生物合成和种子萌发, 在AtTudor2 T-DNA插入突变体和AtTudor2 RNAi转基因株系中, 用于GA生物合成的关键酶AtGA20ox3的表达明显下调[46], 由此推测在油菜GA生物合成过程中起调控作用进而影响一次有效分枝高度。对L-半乳糖-1,4-内酯(L-GalL)表现特异性应答, 其突变体表现根短且莲座小,也对细胞壁的合成和延伸有影响[47]。编码衰老相关的枯草蛋白酶,突变体的花序分枝和角果数量增多。可能参与细胞外膜或质膜的靶向定位或释放质外体信号, 并且在生殖发育和衰老的进程中抑制花序分枝和角果的产生, 从而最大限度地减少分枝和果实间对资源的竞争[48], 同时, 在生长后期的拟南芥茎中它被鉴定出是一种细胞壁蛋白, 而且是蛋白酶抑制剂[49]。编码一种富含亮氨酸的重复蛋白, 该蛋白直接与MADS结构域转录因子相互作用[50]。在诱导开花时, 茎尖分生组织转化为花序分生组织, 其表达增加并调控开花时间[51]; 从诱导花到心皮和雄蕊成熟的过程中,也在特定的细胞中发挥重要作用, 它可能成为联合MADS结构域转录因子在生长发育调控和信号传导中发挥重要作用[52]。本研究探究了株高和一次有效分枝高度对产量的影响并且挖掘出可能与油菜株高和一次有效分枝高度相关的候选基因, 为油菜株型改良以及后续基因的研究提供理论依据, 为下一步的功能验证奠定基础。
4 结论
结合遗传连锁定位和全基因组关联分析, 在A02、A03、A04、A07、C02、C06鉴定出与株高相关的位点, 并筛选出、等候选基因; 在A01、A02、A09、C02找到与一次有效分枝高度相关的位点, 筛选出、等候选基因, 并验证了部分基因在极端材料中的表达情况。
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops. org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuo wxb.aspx。
[1] Wang B, Smith S M, Li J. Genetic regulation of shoot architecture., 2018, 69: 437–468.
[2] Cai G, Yang Q, Chen H, Yang Q, Zhang C, Fan C, Zhou Y. Genetic dissection of plant architecture and yield-related traits in., 2016, 6: 21625.
[3] Wang Y, Li J. Genes controlling plant architecture., 2006, 17: 123–129.
[4] Liu C, Wang J, Huang T, Wang F, Yuan F, Cheng X, Zhang Y, Shi S, Wu J, Liu K. A missense mutation in the VHYNP motif of a DELLA protein causes a semi-dwarf mutant phenotype in., 2010, 121: 249–258.
[5] Khush G S. Green revolution: the way forward., 2001, 2: 815.
[6] Dill A, Jung H S, Sun T P. The DELLA motif is essential for gibberellin-induced degradation of RGA., 2001, 98: 14162–14167.
[7] Peng J, Carol P, Richards D E, King K E, Cowling R J, Murphy G P, Harberd N P. Thegene defines a signaling pathway that negatively regulates gibberellin responses., 1997, 11: 3194–3205.
[8] Rieu I, Ruiz-Rivero O, Fernandez-Garcia N, Griffiths J, Powers S J, Gong F, Linhartova T, Eriksson S, Nilsson O, Thomas S G, Phillips A L, Hedden P. The gibberellin biosynthetic genes AtGA20ox1 and AtGA20ox2 act, partially redundantly, to promote growth and development throughout thelife cycle., 2008, 53: 488–504.
[9] Doebley J, Stec A, Hubbard L. The evolution of apical dominance in maize., 1997, 386: 485–488.
[10] Lewis J M, Mackintosh C A, Shin S, Gilding E, Kravchenko S, Baldridge G, Zeyen R, Muehlbauer G J. Overexpression of the maizegene in wheat suppresses tiller development., 2008, 27: 1217–1225.
[11] Schumacher K, Schmitt T, Rossberg M, Schmitz G, Theres K. The Lateral suppressor () gene of tomato encodes a new member of the VHIID protein family., 1999, 96: 290–295.
[12] Long J, Barton M K. Initiation of axillary and floral meristems in Arabidopsis., 2000, 218: 341–353.
[13] Li X, Qian Q, Fu Z, Wang Y, Xiong G, Zeng D, Wang X, Liu X, Teng S, Hiroshi F, Yuan M, Luo D, Han B, Li J. Control of tillering in rice., 2003, 422: 618–621.
[14] Schmitz G, Tillmann E, Carriero F, Fiore C, Cellini F, Theres K. The tomato Blind gene encodes a MYB transcription factor that controls the formation of lateral meristems., 2002, 99: 1064–1069.
[15] 付正莉, 刘蕊, 王宁宁, 朱克明, 陈松, 张洁夫, 谭小力. 植物分枝发育调控的研究进展. 江苏农业科学, 2018, 46(13): 17–21. Fu Z L, Liu R, Wang N N, Zhu K M, Chen S, Zhang J F, Tan X L. Advances in research on regulation of plant branch development., 2018, 46(13): 17–21 (in Chinese).
[16] Li H, Li J, Song J, Zhao B, Guo C, Wang B, Zhang Q, Wang J, King G J, Liu K. An auxin signaling gene BnaA3.IAA7 contributes to improved plant architecture and yield heterosis in rapeseed., 2018, 222: 837–851.
[17] Han K, Lee H Y, Ro N Y, Hur O S, Lee J H, Kwon J K, Kang B C. QTL mapping and GWAS reveal candidate genes controlling capsaicinoid content in., 2018, 16: 1546–1558.
[18] 王嘉, 荆凌云, 荐红举, 曲存民, 谌利, 李加纳, 刘列钊. 甘蓝型油菜株高、第一分枝高和分枝数的QTL检测及候选基因筛选. 作物学报, 2018, 41: 1027–1038. Wang J, Jing L Y, Jian H J, Qu C M, Chen L, Li J N, Liu L Z. Quantitative trait loci mapping for plant height, the first branch height, and branch number and possible candidate genes screening inL., 2018, 41: 1027–1038 (in Chinese with English abstract).
[19] Zhao W, Wang X, Wang H, Tian J, Li B, Chen L, Chao H, Long Y, Xiang J, Gan J, Liang W, Li M. Genome-wide identification of QTL for seed yield and yield-related traits and construction of a high-density consensus map for QTL comparison in, 2016, 7: 17.
[20] Luo Z, Wang M, Long Y, Huang Y, Shi L, Zhang C, Liu X, Fitt B D L, Xiang J, Mason A S, Snowdon R J, Liu P, Meng J, Zou J. Incorporating pleiotropic quantitative trait loci in dissection of complex traits: seed yield in rapeseed as an example., 2018, 130: 1569–1585.
[21] 贺亚军, 吴道明, 傅鹰, 钱伟. 利用DH和IF2群体检测甘蓝型油菜株高相关性状QTL. 作物学报, 2018, 44: 533–541. He Y J, Wu D M, Fu Y, Qian W. Detection of QTLs for plant height related traits inL. using DH and immortalized F2population., 2018, 44: 533–541 (in Chinese with English abstract).
[22] Shen Y, Xiang Y, Xu E, Ge X, Li Z. Major co-localized QTL for plant height, branch initiation height, stem diameter, and flowering time in an alien introgression derivedDH population., 2018, 9: 390.
[23] Zheng M, Peng C, Liu H, Tang M, Yang H, Li X, Liu J, Sun X, Wang X, Xu J, Hua W, Wang H. Genome-wide association study reveals candidate genes for control of plant height, branch initiation height and branch number in rapeseed (L)., 2017, 8: 1246.
[24] Sun C, Wang B, Yan L, Hu K, Liu S, Zhou Y, Guan C, Zhang Z, Li J, Zhang J, Chen S, Wen J, Ma C, Tu J, Shen J, Fu T, Yi B. Genome-wide association study provides insight into the genetic control of plant height in rapeseed (L)., 2016, 7: 1102.
[25] Li F, Chen B, Xu K, Gao G, Yan G, Qiao J, Li J, Li H, Li L, Xiao X, Zhang T, Nishio T, Wu X. A genome-wide association study of plant height and primary branch number in rapeseed ()., 2016, 242: 169–177.
[26] Lu K, Wei L, Li X, Wang Y, Wu J, Liu M, Zhang C, Chen Z, Xiao Z, Jian H, Cheng F, Zhang K, Du H, Cheng X, Qu C, Qian W, Liu L, Wang R, Zou Q, Ying J, Xu X, Mei J, Liang Y, Chai Y R, Tang Z, Wan H, Ni Y, He Y, Lin N, Fan Y, Sun W, Li N N, Zhou G, Zheng H, Wang X, Paterson A H, Li J. Whole-genome resequencing revealsorigin and genetic loci involved in its improvement., 2019, 10: 1154.
[27] Wang S, Basten C, Zeng Z. Windows QTL Cartographer v2.5. Department of statistics, North Carolina State University, 2007, Raleigh, N C.
[28] McCouch S R, Cho Y G, Yano M, Paul E, Blinstrub M, Morishima H, Kinoshita T. Report on QTL nomenclature., 1997, 14: 11–13.
[29] Yu J, Pressoir G, Briggs W H, Bi I V, Yamasaki M, Doebley J F, McMullen M D, Gaut B S, Nielsen D M, Holland J B. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness., 2006, 38: 203–208.
[30] Lu K, Xiao Z, Jian H, Peng L, Qu C, Fu M, He B, Tie L, Liang Y, Xu X, Li J. A combination of genome-wide association and transcriptome analysis reveals candidate genes controlling harvest index-related traits in., 2016, 6: 36452.
[31] Chalhoub B, Denoeud F, Liu S, Parkin I A P, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, Corréa M, Da Silva C, Just J, Falentin C, Koh C S, Le Clainche I, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao M, Edger P P, Chelaifa H, Tack D, Lassalle G, Mestiri I, Schnel N, Le Paslier M C, Fan G, Renault V, Bayer P E, Golicz A A, Manoli S, Lee T H, Thi V H D, Chalabi S, Hu Q, Fan C, Tollenaere R, Lu Y, Battail C, Shen J, Sidebottom C H D, Wang X, Canaguier A, Chauveau A, Bérard A, Deniot G, Guan M, Liu Z, Sun F, Lim Y P, Lyons E, Town C D, Bancroft I, Wang X, Meng J, Ma J, Pires J C, King G J, Brunel D, Delourme R, Renard M, Aury J M, Adams K L, Batley J, Snowdon R J, Tost J, Edwards D, Zhou Y, Hua W, Sharpe A G, Paterson A H, Guan C, Wincker P. Early allopolyploid evolution in the post-Neolithicoilseed genome., 2014, 345: 950–953.
[32] Raboanatahiry N, Chao H, Dalin H, Pu S, Yan W, Yu L, Wang B, Li M. QTL alignment for seed yield and yield related traits in., 1997, 9: 1127.
[33] Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J. Unraveling the complex trait of crop yield with quantitative trait loci mapping in., 2009, 182: 851–861.
[34] Schultz C J, Johnson K L, Currie G, Bacic A. The classical arabinogalactan protein gene family of., 2000, 12: 1751–1768.
[35] 谢田田, 陈玉波, 黄吉祥, 张尧锋, 徐爱遐, 陈飞, 倪西源, 赵坚义. 甘蓝型油菜不同发育时期株高QTL的动态分析. 作物学报, 2012, 38: 1802–1809. Xie T T, Chen Y B, Huang J X, Zhang Y F, Xu A X, Chen F, Ni X Y, Zhao J Y. Dynamic analysis of QTL for plant height of rapeseed at different developmental stages., 2012, 38: 1802–1809 (in Chinese with English abstract).
[36] Goda H, Sawa S, Asami T, Fujioka S, Shimada Y, Yoshida S. Comprehensive comparison of Auxin-regulated and brassinosteroid-regulated genes in Arabidopsis., 1997, 134: 1555–1573.
[37] Lee D J, Park J W, Lee H W, Kim J. Genome-wide analysis of the auxin-responsive transcriptome downstream ofand its expression analysis reveal the diversity and complexity of auxin-regulated gene expression., 2009, 60: 3935–3957.
[38] Redman J C, Haas B J, Tanimoto G, Town C D. Development and evaluation of anwhole genome Affymetrix probe array., 2004, 38: 545–561.
[39] Kim D W, Jeon S J, Hwang S M, Hong J C, Bahk J D. The C3H-type zinc finger protein GDS1/C3H42 is a nuclear-speckle-localized protein that is essential for normal growth and development in., 2016, 250: 141–153.
[40] Deeken R, Engelmann J C, Efetova M, Czirjak T, Muller T, Kaiser W M, Tietz O, Krischke M, Mueller M J, Palme K, Dandekar T, Hedrich R. An integrated view of gene expression and solute profiles oftumors: a genome-wide approach., 2006, 18: 3617–3634.
[41] Shani Z, Dekel M, Roiz L, Horowitz M, Kolosovski N, Lapidot S, Alkan S, Koltai H, Tsabary G, Goren R, Shoseyov O. Expression of endo-1,4-beta-glucanase (cel1) inis associated with plant growth, xylem development and cell wall thickening., 2006, 25: 1067–1074.
[42] Xiong J, Cui X, Yuan X, Yu X, Sun J, Gong Q. The Hippo/STE20 homolog SIK1 interacts with MOB1 to regulate cell proliferation and cell expansion in., 2016, 67: 1461–1475.
[43] Wang Z, Chen F, Li X, Cao H, Ding M, Zhang C, Zuo J, Xu C, Xu J, Deng X, Xiang Y, Soppe W J J, Liu Y. Arabidopsis seed germination speed is controlled by SNL histone deacetylase-binding factor-mediated regulation of AUX1., 2016, 7: 13412.
[44] Rashotte A M, Carson S D, To J P, Kieber J J. Expression profiling of cytokinin action in Arabidopsis., 2003, 132: 1998–2011.
[45] Hanzawa Y, Imai A, Michael A J, Komeda Y, Takahashi T. Characterization of the spermidine synthase-related gene family in., 2002, 527: 176–180.
[46] Liu S, Jia J, Gao Y, Zhang B, Han Y. The AtTudor2, a protein with SN-Tudor domains, is involved in control of seed germination in., 2010, 232: 197–207.
[47] Gao Y, Badejo A A, Sawa Y, Ishikawa T. Analysis of two L-galactono-1,4-lactone-responsive genes with complementary expression during the development of., 2012, 53: 592–601.
[48] Martinez D E, Borniego M L, Battchikova N, Aro E M, Tyystjarvi E, Guiamet J J. SASP, a Senescence-Associated Subtilisin Protease, is involved in reproductive development and determination of silique number in., 2015, 66: 161–174.
[49] Wei H, Brunecky R, Donohoe B S, Ding S Y, Ciesielski P N, Yang S, Tucker M P, Himmel M E. Identifying the ionically bound cell wall and intracellular glycoside hydrolases in late growth stage Arabidopsis stems: implications for the genetic engineering of bioenergy crops., 2015, 6: 315.
[50] Gamboa A, Paez-Valencia J, Acevedo G F, Vazquez-Moreno L, Alvarez-Buylla R E. Floral transcription factor AGAMOUS interacts in vitro with a leucine-rich repeat and an acid phosphatase protein complex., 2001, 288: 1018–1026.
[51] Torti S, Fornara F, Vincent C, Andres F, Nordstrom K, Gobel U, Knoll D, Schoof H, Coupland G. Analysis of the Arabidopsis shoot meristem transcriptome during floral transition identifies distinct regulatory patterns and a leucine-rich repeat protein that promotes flowering., 2012, 24: 444–462.
[52] Acevedo F G, Gamboa A, Paéz-Valencia J, Jiménez-Garcı́a L F, Izaguirre-Sierra M, Alvarez-Buylla E R. FLOR1, a putative interaction partner of the floral homeotic protein AGAMOUS is a plant-specific intracellular LRR., 2004, 167: 225–231.
Candidate genes screening for plant height and the first branch height based on QTL mapping and genome-wide association study in rapessed (L.)
HUO Qiang1,2,**, YANG Hong1,2,**, CHEN Zhi-You1,2, JIAN Hong-Ju1,2, QU Cun-Min1,2, LU Kun1,2, and LI Jia-Na1,2,*
1College of Agronomy and Biotechnology, Southwest University / Chongqing Engineering Research Center for Rapeseed, Chongqing 400715, China;2Academy of Agricultural Sciences, Southwest University, Chongqing 400715, China
Plant height (PH) and the first branch height (BH) are important agronomic traits closely related to thickness of pod canopy and harvest index of. There are many reports on quantitative trait locus (QTL) and genome-wide association study (GWAS) for PH, but few reports on QTL and GWAS localization and candidate gene screening for BH. In this study, QTLs for PH and BH using the 186 recombinant inbred lines (RIL) in the two environments of 2016 and 2017 were detected based on the high-density genetic linkage map. A total of eight PH QTLs located on chromosomes A03, A04, and A09 were detected in the two environments of 2016 and 2019, with the explained phenotypic variation of individual QTL range from 4.60% to 13.29%, among which overlapped QTLs (q-2017PH-A04-2 and q-BLUP-PH-A04-2) were detected both in 2017 and based on Best Linear Unbiased Prediction (BLUP). Nine BH QTLs located on chromosomes A01, A02, A05, A09, C01, and C05 were detected, in which a single QTL explained 5.12% to 19.10% of phenotypic variation. Among them, q-2017BH-A09-1, q-BLUP-BH-A09-2, and q-BLUP-BH-A09-3 were overlapped. GWAS for PH and BH was performed using 588 resequencing natural populations established by our previous study. A total of 50 SNPs associated with PH and 12 SNPs associated with BH were detected in two years. Thirteen PH candidate genes involved in cell proliferation, cell multiplication, cell cycle and cell wall activity and eleven BH candidate genes involved in the synthesis and metabolism of gibberellin and spermidine, ribosome composition, photosynthesis and germination were screened based on the physical locations of SNPs, and their expressions in extreme phenotypes by qRT-PCR. This result will provide a theoretical basis for improving plant architecture and subsequent functional studies of genes.
; plant height; first branch height; gene localization; candidate gene screening
本研究由重庆市民生工程主题专项(cstc2016shms-ztzx80020), 国家自然科学基金-云南联合基金项目(U1302266)和国家重点研发计划项目(2018YFD0100504-05)资助。
This study was supported by the Special Project of Chongqing People’s Livelihood (cstc2016shms-ztzx80020), the National-Yunnan United Natural Science Foundation of China (U1302266), and the National Key Research and Development Plan (2018YFD0100504-05).
10.3724/SP.J.1006.2020.94067
李加纳, E-mail: ljn1950@swu.edu.cn, Tel: 023-68250701
**同等贡献(Contributed equally to this work)
霍强, E-mail: 354011524@qq.com; 杨鸿, E-mail: 583791495 @qq.com
2019-04-28;
2019-08-09;
2019-09-02.
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20190902.1554.006.html
