小麦品种扬麦16赤霉病抗扩展QTL定位及分析
2020-01-02胡文静陆成彬王凤菊刘金栋蒋正宁王金平朱展望徐小婷郝元峰何中虎高德荣
胡文静 张 勇 陆成彬 王凤菊 刘金栋 蒋正宁 王金平朱展望 徐小婷 郝元峰 何中虎,3 高德荣,*
小麦品种扬麦16赤霉病抗扩展QTL定位及分析
胡文静1张 勇1陆成彬1王凤菊2刘金栋2蒋正宁1王金平2朱展望2徐小婷2郝元峰2何中虎2,3高德荣1,*
1江苏里下河地区农业科学研究所/ 农业农村部长江中下游小麦生物学与遗传育种重点实验室, 江苏扬州 225007;2国家小麦改良中心 / 中国农业科学院作物科学研究所, 北京 100081;3国际玉米小麦改良中心(CIMMYT)中国办事处, 北京 100081
扬麦系列品种赤霉病抗性在世界范围内得到重视, 但其抗性遗传机制尚不清楚。扬麦16是近年来大面积推广的抗赤霉病品种, 本研究以扬麦16与中麦895杂交构建的174个双单倍体(double haploid lines, DH)系为材料, 于2017—2019年连续3年对该群体采用单花滴注进行赤霉病抗扩展鉴定。利用660K SNP芯片构建高密度遗传图谱, 共检测到6个抗性QTL, 分别位于2DL、3BL、4BS、4DS、5BL和6AS染色体上。除4BS位点外, 其他5个抗性等位基因均来源于扬麦16。和均在多年被检测到, 可解释8.8%~15.0%的表型变异;、仅在1年被检测到, 分别解释10.5%和14.7%的表型变异;和来源于中麦895的仅在1年被检测到且效应仅为6.4%和8.3%。QTL效应分析结果表明, 相较于单个位点, 多个抗性QTL的聚合可显著降低赤霉病严重度。扬麦16抗赤霉病QTL将为揭示扬麦品种抗性遗传机制及开发相应分子标记奠定基础。
小麦; 赤霉病; QTL; 标记辅助育种
小麦赤霉病(Wheat scab, Fusarium head blight, FHB)是由禾谷镰刀菌()等引起的一种世界性真菌病害[1]。我国长江中下游麦区是赤霉病的常发区和重发区, 2012—2015年, 江苏省年均赤霉病发生面积约120万公顷[2]。近年来, 随着气候变暖和玉米秸秆还田, 小麦赤霉病发生愈来愈重, 正迅速扩展到黄淮麦区。据统计, 近10年, 河南省赤霉病年均发生面积达110万公顷左右, 其中2012年达333万公顷, 2016年达117万公顷[3]。
赤霉病的侵染时期主要是小麦开花期, 赤霉菌侵染小花后迅速在穗部扩展, 在小麦籽粒灌浆成熟过程中不断繁殖, 进而积累各种毒素, 例如脱氧雪腐镰孢菌烯醇(deoxynivalenol, DON)、雪腐镰孢菌烯醇(nivalenol, NIV)和玉米赤霉烯酮(zearalenol, ZEN), 严重影响小麦产量, 并对人畜健康造成巨大伤害, 成为粮食安全的主要威胁[4]。选育和种植抗病品种是应对赤霉病危害最经济、安全的途径。小麦赤霉病的抗性主要可以分为抗侵染(type I)、抗扩展(type II)、抗DON积累(type III)和籽粒抗性(type IV) 4种类型, 其中type II类型研究最深入[5]。多年来, 已报道超过250个抗赤霉病QTL, 覆盖小麦全部21条染色体[6], 但目前明确的小麦抗赤霉病基因只有7个, 其中和是抗扩展类型,和是抗侵染类型, 仅已经被克隆[7-8]。苏麦3号是国际上研究和利用最广泛的赤霉病抗源, 望水白是已知的携带抗赤霉病位点最多的小麦品种之一[3]。它们均携带主效抗病基因, 但两者综合农艺性状较差, 秆高易倒伏、穗数和穗粒数少、粒重低, 虽利用它们选育出一批抗性好的材料, 例如宁7840等, 但因其综合丰产性差未得到大面积应用[9]。据报道, 聚合和其他抗赤霉病位点, 可累加抗性效应, 提高抗性水平[10-11]。扬麦系列品种一直是长江中下游麦区主体品种, 多数品种表现中抗赤霉病。扬麦16是近年来长江中下游推广面积最大的小麦品种, 利用其为亲本选育出优质丰产抗赤霉病的小麦品种如扬麦23和扬麦28等, 表明扬麦16在小麦遗传改良中可以作为稳定的抗赤亲本使用。经中国农业科学院开发的小麦功能基因标记检测, 扬麦16不含[12-13], 可能存在其他主效抗赤霉病基因。因此, 挖掘扬麦16的抗赤霉病基因, 拓宽小麦赤霉病抗源, 对小麦抗赤霉病育种具有重要意义。本研究利用来自扬麦16/中麦895 的174份双单倍体家系, 经660K SNP芯片检测, 构建高密度遗传图谱, 结合多年表型鉴定结果, 挖掘扬麦16抗赤霉病QTL, 为开展标记辅助抗赤霉病育种提供技术支撑。
1 材料与方法
1.1 试验材料及田间试验
以扬麦16 (Yangmai 16, 简称YM16, 系谱: 扬91F138 (扬麦158选系)/扬90-30)为母本, 中麦895 (Zhongmai 895, 简称ZM895, 系谱: 周麦16/荔垦4号)为父本杂交, 利用玉米花粉诱导双单倍体培育174份DH家系。以苏麦3号(Sumai 3, 简称SM3)和周麦18 (Zhoumai 18, 简称ZM18)为抗赤霉病和感赤霉病对照。2016—2017年度(简称为2017年)于国际玉米小麦改良中心(International Maize and Wheat Improvement Center, CIMMYT)温室种植, 花盆直径20 cm, 每盆播种8粒, 最后定苗至5株, 2次重复, 参照CIMMYT的正常管理方法管理温室。2017—2018年度和2018—2019年度(简称为2018年和2019年)于江苏里下河地区农业科学研究所基地种植, 采用完全随机设计试验, 2行区, 每行30粒, 2次重复, 行长1.3 m, 行距0.3 m, 参照当地的正常栽培方法管理大田。
1.2 赤霉病鉴定和表型数据处理
参照 Yu 等[14]方法制备赤霉菌孢子悬浮液(5000孢子 mL–1), 采用单花滴注法接种。于小麦开花初期, 用注射器吸取10 µL孢子液注射入麦穗中间小穗的一个小花中(顶部小穗开始自上而下第6个小穗), 接种每系15个穗子。温室单花滴注接种后套袋保湿3 d, 田间单花接种后立即采用人工弥雾保湿(每半小时田间弥雾喷水5 min, 6:00–18:00)。接种21 d后调查病小穗数, 计算病小穗率。病小穗率(percentage of scabbed spikelets, 简称PSS)作为赤霉病严重度的衡量指标[9]。病小穗率(%) = (病小穗数/总小穗数) ×100%。用 DPS 软件和IciMapping的ANOVA功能进行群体表型数据的相关分析、方差分析及遗传力的估算。
1.3 分子标记的检测
取小麦幼苗, 采用CTAB法提取基因组DNA[15], 利用Affymetrix SNP技术检测平台(北京博奥生物有限公司)小麦660K SNP芯片分析亲本和群体, 利用Genomestudio v1.0软件进行多态性分析和整合(Illumina, http://www.illumina.com/)。将13个包括和[12]等重要农艺性状功能基因的相应KASP标记也整合入多态性标记中。
1.4 遗传图谱构建和QTL定位
筛选出缺失率<10%, 最小等位基因频率>30%的多态性标记, 使用IciMapping v4.1 (http://www. isbreeding.net/)的BIN功能进行去冗余。根据SNP侧翼序列比对到中国春参考基因组的物理位置(IWGSC RefSeq v1.0, https://wheat-urgi.versailles. inra.fr/)和SNP在小麦660K整合图谱(赵光耀, 私人通讯) KJ-RIL遗传图谱中的位置[16], 挑选461个分布在小麦21条染色体的SNP作为Anchor标记, 利用IciMapping v4.1的MAP功能将标记分组, 利用MSTmap(http://mstmap.org/)的Kosambi功能对各组标记排序和计算遗传距离。利用JoinMap v4.0[17]校正遗传长度, 得到最终遗传图谱。利用MapChart 2.3 (https://www.wur.nl/en/show/Mapchart.htm)绘制遗传图谱。利用IciMapping v4.1的完备区间作图法(Inclusive composite interval mapping, ICIM)检测抗赤霉病QTL, LOD阈值设为2.5[18]。将同一染色体上检测到的峰值所在遗传位置之间距离小于10 cM的QTL视为同一个位点。QTL的命名方法为“Q”加性状缩写, 加单位名称缩写, 加QTL所在的染色体, 同一染色体上多个QTL用.1、.2、.3、……来表示。例如代表 5A 染色体上检测出来的控制赤霉病抗扩展的第一个QTL。将在2个或者超过2个年份下能够检测到的QTL定义为“稳定QTL”, 将效应值大于10%的QTL定义为“具有较大表型贡献率的QTL”。为了与前人结果比较, 将连锁标记或者基因的序列与中国春参考基因组序列的EnsemblPlants数据库(http://plants.ensembl.org/)比对, 获得标记或者基因的物理位置[19]。
2 结果与分析
2.1 亲本及DH群体的抗性表现
由表1和图1可以看出, 3年试验的PSS范围, 扬麦16是14.2%~23.3%, 中麦895是58.7%~69.7%, 抗病对照苏麦3号是5.2%~6.1%, 感病对照周麦18是60.2%~70.1%。群体的偏度和峰度绝对值均<1, 分布呈连续的正态分布, 可用于QTL定位研究[20]。

图1 扬麦16/中麦895 DH群体赤霉病严重度频次分布(2017–2019)
横坐标代表病小穗率, 纵坐标代表相关病小穗率范围内的家系数目。
The abscissa indicates the percentage of scabbed spikelets, and the ordinate indicates the line number.
3年试验结果均表明双亲赤霉病的病小穗率存在极显著的差异, DH群体赤霉病严重度的最小值和最大值之间差异明显, 存在超亲分离, 可以初步判断双亲都携带抗性基因且位点互补, 通过杂交可以得到超亲分离的株系。抗病和感病对照的病小穗率亦存在极显著差异。
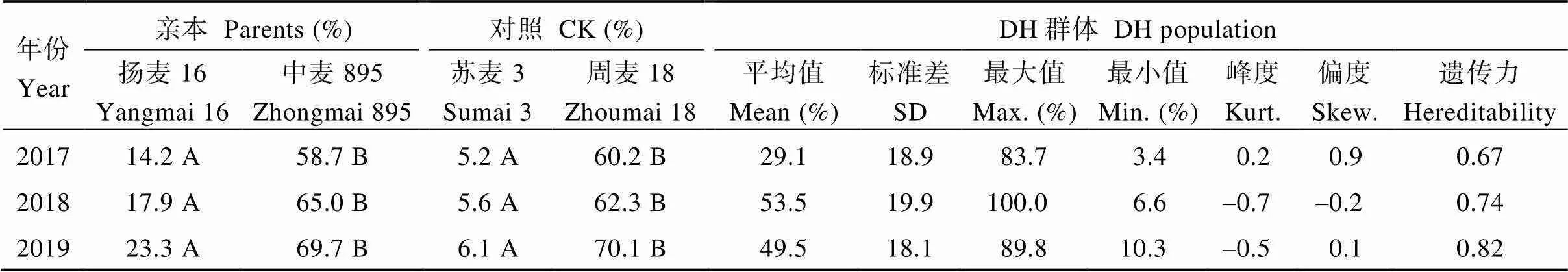
表1 扬麦16/中麦895 DH群体、亲本和对照的赤霉病严重度
数据后不同字母表示不同基因型间赤霉病严重度差异显著(< 0.01)。
Values followed by different letters are significant by different in FHB severity among genotypes (< 0.01).
2.2 遗传连锁图谱
利用660K SNP芯片, 过滤和筛选得到152,310个高质量多态性SNP标记, 经过去冗余分析后, 最终“扬麦16/中麦895”高密度遗传图谱的上图标记为14,480个。图谱覆盖小麦21条染色体, 长度为3681.7 cM, 密度为3.9个标记 cM–1。分布于小麦A、B和D染色体组的标记数分别为5036、7634和1810个, 连锁长度分别为1223.2、1035.6和1423.0 cM。其中, 标记密度最高的是1B染色体, 平均11.7个标记 cM–1, 标记密度最低的是7D染色体, 平均0.8个标记 cM–1。
2.3 抗赤霉病QTL定位
共检测到6个抗赤霉病QTL, 分别位于2DL、3BL、4BS、4DS、5BL和6AS上。除外, 其他5个QTL均来自扬麦16。在3年中均被检测到, 抗性贡献率8.8%~13.7%,可在2年同时被检测到, 抗性贡献率9.5%~15.0%。这2个QTL是本研究检测到的稳定的抗赤霉病位点。和分别在2017和2019年被检测到, 抗性贡献率分别为10.5%和14.7%。和均仅在2018年被检测到, 抗性贡献率分别为8.3%和6.4% (表2和图2)。
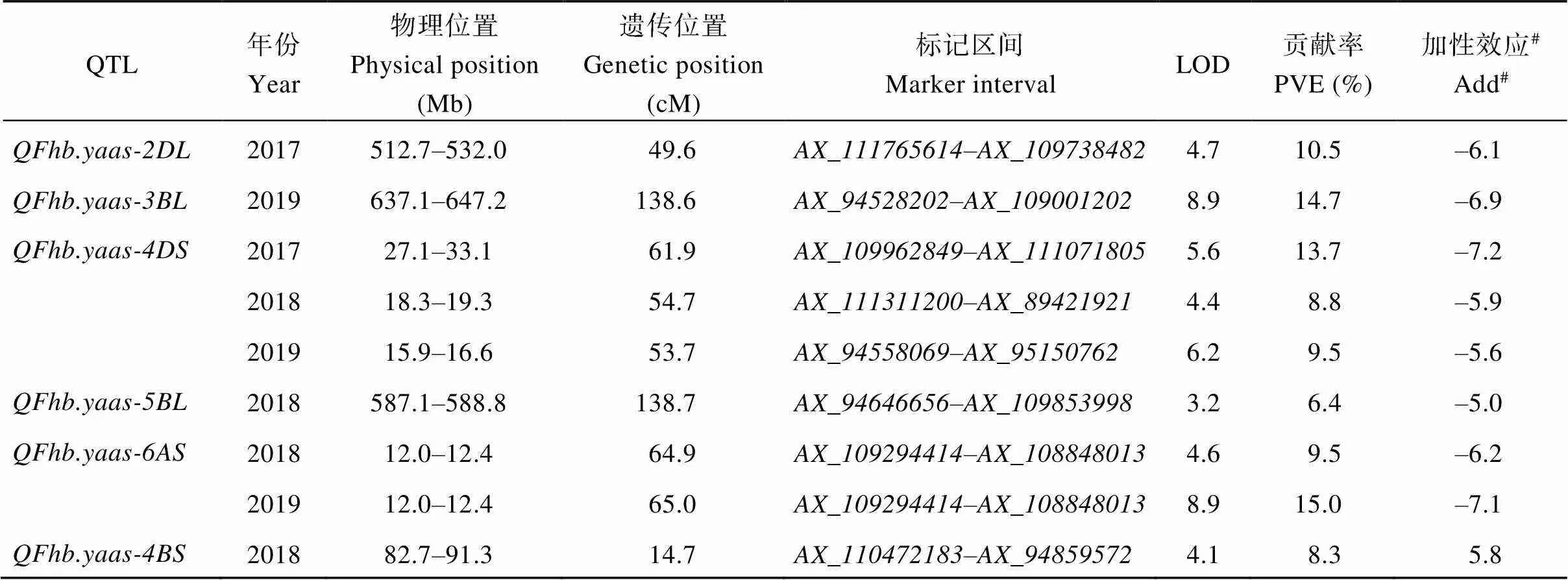
表2 扬麦16/中麦895 DH 群体抗赤霉病QTL定位
#加性效应为负说明赤霉病抗性来源于扬麦16, 加性效应为正说明赤霉病抗性来源于中麦895。
#The resistance alleles at loci with negative additive effects are from YM16 and that with a positive additive effect is from ZM895.

图2 扬麦16/中麦895 DH群体抗赤霉病QTL定位情况
连锁群右边是标记名称, 左边是遗传位置(CM)。红色、蓝色和绿色分别代表2017、2018和2019年定位到的QTL, QTL右边对应LOD值。
Markers’ names are shown to the right of vertical axis, and their genetic positions are shown in cM to the left. Red, blue, and green represent 2017, 2018, and 2019, respectively. LOD values of QTL are shown on the right side.
2.4 QTL效应分析
将在2个或者超过2个环境下能够检测到的QTL定义为“稳定QTL”, 本研究分别在3年和2年环境下均检测到和, 根据它们的基因型将174个家系分为4种类型, 用3年赤霉病严重度平均值进行类型间的差异分析。38个家系同时含有和, 赤霉病严重度范围9.9%~57.4% (平均32.3%), 多数家系PSS小于40%, 仅有8个家系PSS在40%~60%范围内; 28个家系只含有, 赤霉病严重度范围18.9%~78.8% (平均44.8%), 18个家系PSS小于50%, 10个家系PSS在50%~80%范围内; 有62个家系只含有, 赤霉病严重度范围18.6%~71.3% (平均43.2%), 41个家系PSS小于50%, 21个家系PSS在50%~80%范围内; 46个家系不含上述2个QTL, 赤霉病严重度范围26.1%~88.0% (平均54.3%), 多数家系PSS大于40%, 仅有6个家系PSS在20%~40%范围。聚合2个抗性位点的家系比含有1个抗性位点和不含有抗性位点的家系赤霉病严重度显著降低(表3)。
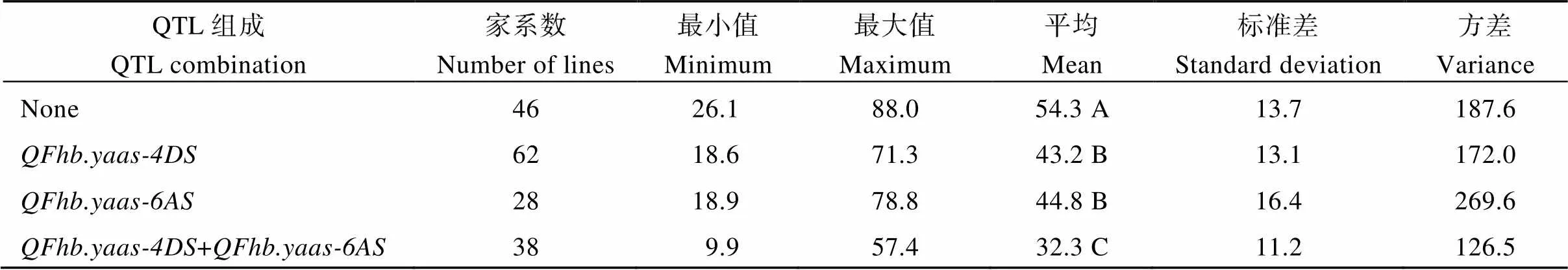
表3 扬麦16/中麦895 DH群体中存在不同稳定赤霉病抗扩展QTL家系的赤霉病严重度情况
数据后不同字母表示不同基因型间赤霉病严重度差异显著(< 0.01)。
Values followed by different letters are significant by different in FHB severity among genotypes (< 0.01).
根据本研究中定位到的效应值大于10%的抗性QTL、、和将174个DH家系分为5种类型, 用3年PSS平均值进行类型间的差异分析。10个家系同时含有4个抗性QTL, 发病较轻, 平均PSS是30.5%; 46个家系含有3个抗性QTL, 平均PSS是36.9%; 65个家系含有2个抗性QTL, 平均PSS是41.8%; 46个家系只含有1个抗性QTL, 平均PSS是53.84%; 7个家系不含有任何一个上述4个抗性QTL, 平均PSS是67.1% (表4和图3)。
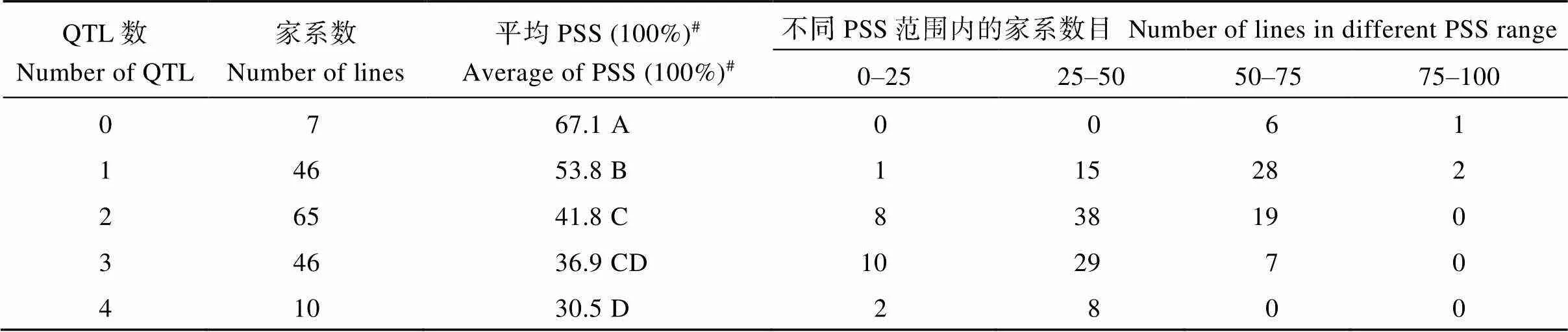
表4 扬麦16/中麦895 DH群体中存在不同赤霉病抗扩展QTL家系的赤霉病严重度情况
PSS (percentage of scabbed spikelets)代表赤霉病严重度。#数据后不同字母表示不同基因型间赤霉病严重度差异极显著(< 0.01)。
PSS (percentage of scabbed spikelets) indicates the FHB severity.#Values followed by different letters are significant by different in FHB severity among genotypes (< 0.01).

图3 DH家系所含QTL数目与其平均赤霉病严重度的关系
PSS (percentage of scabbed spikelets)代表赤霉病严重度。
PSS (percentage of scabbed spikelets) indicates the FHB severity.
3 讨论
3.1 QTL定位结果与已知抗赤霉病QTL/基因比较
在扬麦16/中麦895 DH群体中共检测到6个赤霉病抗扩展QTL, 其中和在多年均可被检测到, 是来源于扬麦16的稳定QTL位点。根据比对结果,与紧密连锁, Srinivasachary等[21]报道附近确实存在赤霉病抗侵染QTL, 来源于Spark,可显著降低小麦对赤霉病抗侵染能力, 但其与赤霉病抗扩展性不相关[22], Buerstmayr等[23]研究表明携带位点的小麦品种易感染赤霉病, 表明较高的株高营造的穗部局部低湿度环境可以减少赤霉病孢子侵染小麦。本研究采用一年CIMMYT温室单花滴注然后套袋保湿, 两年扬州田间单花滴注试验采取弥雾保湿, 株高对穗部的微环境存在一定的影响, 从而影响发病的严重度。将2017—2019年DH群体株高数据平均数(未发表资料)与赤霉病严重度平均值进行相关性分析, 发现株高均值与赤霉病严重度均值相关系数–0.33 (<0.01), 表明该群体赤霉病抗性与株高存在一定相关性。群体中携带抗病等位基因家系的平均株高显著高于携带感病等位基因家系的平均株高(< 0.01), 表明该位点确实与株高显著相关。该位点为“一因多效”或者“紧密连锁”, 可通过后续增加温室环境下单花滴注试验进行验证。另据报道, 小麦粒重基因启动子区域的单倍型变异除了与粒重相关外, 还与株高显著相关[24], 本研究定位到了位于6AS染色体上的抗赤霉病QTL, DH群体2017—2019年的株高平均值(未发表资料)与赤霉病严重度平均值相关系数为–0.33 (<0.01), 表明该位点的赤霉病抗性与株高也存在一定的关系。
与来源于扬麦13的位置相近[6]。扬麦13的系谱是扬88-84//Maris Dove/扬麦3号, 扬麦3号是阿夫的红壳选系; 扬麦16的系谱是扬麦158优系(91F138)/扬90-30, 扬麦158的系谱是扬麦4号//St1472/506, 扬麦4号的系谱是南大2419/胜利麦1-3-2//阿夫选系。由于扬麦品种的亲本中存在共同的亲本阿夫, 且亲缘关系较近, 推测与可能是相同抗病位点, 后续我们会继续研究上述材料, 以确定的来源。
其他QTL仅在1年被检测到, 其中和的效应值较大, 来自扬麦16。可解释表型变异10.5%, 经两侧标记序列与中国春参考基因组序列比对发现, 其物理位置区间是512.7~532.0 Mb, 与已报道的Wuhan-1中检测到的2DL上抗赤霉病位点位置相近(513.1 Mb)[25], Wuhan-1与扬麦16均来自长江中下游麦区, 推测可能是相同的QTL。能解释表型变异14.7%, 经比对发现无同位置抗赤霉病QTL报道, 推测可能为新的抗赤霉病位点。小麦赤霉病抗性属于数量性状, 发病受环境影响很大,还需增加多环境试验验证上述QTL的有效性和稳定性。
3.2 抗性改良与QTL/基因聚合
DH家系中仅存在单个抗赤霉病位点时, 多数家系的PSS在50%以上, 但当存在2位点或者2位点以上的QTL聚合时, 平均PSS明显降低。聚合包含4个QTL的家系具有最好的赤霉病抗性, 其中2个家系的PSS在0~25%区间, 达到高抗水平, 其余8个家系的PSS在25%~50%区间, 达到中抗水平, 表明多个抗病基因聚合能表现出较好的抗性。Jia等[10]报道与其他赤霉病抗性QTL/基因的聚合能有效提高小麦的赤霉病抗扩展和抗侵染水平。Arruda等[11]研究表明, 聚合多个3B上抗性位点能使赤霉病抗性水平提高。因此, 将多个抗病基因聚合到同一品种(系)中, 对于提高赤霉病抗性具有重要意义。
3.3 抗赤霉病基因的利用
我国长江中下游麦区是赤霉病的常发区和重发区, 国内外广泛使用的主效抗病基因区段与不利农艺性状存在连锁[26], 因此单纯利用苏麦3号和望水白背景的不足以解决该麦区赤霉病的危害。关于这一点程顺和等[27]早有阐述, 并提出抗赤霉病育种的第2条路线, 即选用丰产、中感-中抗材料杂交, 后代注重丰产, 兼顾抗赤性等选择, 选育出扬麦158、扬麦11、扬麦16等一批抗赤性与丰产性相结合的大面积种植品种。扬麦系列多数品种的赤霉病抗性好且遗传力较强正在被大范围用作育种亲本, 更有价值的是其抗病基因不是, 需要进一步发掘其中的主效赤霉病抗性QTL并开发相应的标记, 为扬麦抗赤霉病基因的高效利用奠定基础。由于多数抗病扬麦品种不具, 将导入扬麦背景, 提高抗赤霉病等级, 不失为解决长江中下游麦区赤霉病危害的有效途径, 如扬16-157 (含), 其在2017—2019年度国家小麦良种重大联合攻关试验中表现高抗赤霉病, 与苏麦3号相当, 2017—2018年品比试验中产量为6325.5 kg hm–2, 比对照扬麦20增产7.0%, 2018—2019年品比试验中产量为6886.5 kg hm–2, 比对照增产4.5%。
4 结论
扬麦16中抗赤霉病, 具有主效QTL, 其中4个QTL具有较大的表型贡献率, 2个QTL多年被检测到, QTL聚合能显著降低小麦赤霉病严重度。扬麦系列小麦品种QTL的发掘可以为小麦抗赤霉病改良提供优异基因资源。
[1] Buerstmayr H, Ban T, Anderson J A. QTL mapping and marker-assisted selection forhead blight resistance in wheat: a review., 2009, 128: 1–26.
[2] 吴佳文, 杨荣明, 朱凤, 田子华. 2015 年江苏省小麦赤霉病发生特点与防控对策探讨. 中国植保导刊, 2016, 36(10): 31–34. Wu J W, Yang R M, Zhu F, Tian Z H. The epidemic characteristics of wheathead blight in Jiangsu province in 2015 and discussion of its control measures., 2016, 36(10): 31–34 (in Chinese).
[3] 张爱民, 阳文龙, 李欣, 孙家柱. 小麦抗赤霉病研究现状与展望. 遗传, 2018, 40: 858–873. Zhang A M, Yang W L, Li X, Sun J Z. Current status and perspective on research againsthead blight in wheat., 2018, 40: 858–873 (in Chinese with English abstract).
[4] 史建荣, 刘馨, 仇剑波, 祭芳, 徐剑宏, 董飞, 殷宪超, 冉军舰. 小麦中镰刀菌毒素脱氧雪腐镰刀菌烯醇污染现状与防控研究进展. 中国农业科学, 2014, 47: 3641–3654. Shi J R, Liu X, Qiu B, Ji F, Xu J H, Dong F, Yin X C, Ran J J. Deoxynivalenol contamination in wheat and its management., 2014, 47: 3641–3654 (in Chinese with English abstract).
[5] Ren J D, Wang Z, Du Z Y, Chen M Z, Zhang Y B, Quan W, Wang Y J, Jiang X, Zhang Z J. Detection and validation of a novel major QTL for resistance tohead blight fromin the terminal region of chromosome 7DL., 2019, 132: 241–255.
[6] Yi X, Cheng J, Jiang Z, Hu W, Bie T, Gao D, Li D, Wu R, Li Y, Chen S, Cheng X, Liu J, Zhang Y, Cheng S. Genetic analysis ofhead blight resistance in CIMMYT bread wheat line C615 using traditional and conditional QTL mapping., 2018, 9: 573. https://doi.org/10.3389/ fpls.2018.00573.
[7] Su Z Q, Bernardo A, Tian B, Chen H, Wang S, Ma H X, Cai S B, Liu D T, Zhang D D, Li T, Trick H, St Amand P, Yu J M, Zhang Z Y, Bai G H. A deletion mutation inconfersresistance tohead blight in wheat., 2019, https://doi.org/10.1038/s41588-019-0425-8.
[8] Li G Q, Zhou J Y, Jia H Y, Gao Z X, Fan M, Luo Y J, Zhao P T, Xue S L, Li N, Yuan Y, Ma S W, Kong Z X, Jia L, An X, Jiang G, Liu W X, Cao W J, Zhang R R, Fan J C, Xu X W, Liu Y F, Kong Q Q, Zheng S H, Wang Y, Qin B, Cao S Y, Ding Y X, Shi J X, Yan H S, Wang X, Ran C F, Ma Z Q. Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance tohead blight., 2019, https://doi.org/10.1038/s41588-019-0426-7.
[9] Li T, Luo M, Zhang D, Wu D, Li L, Bai G. Effective marker alleles associated with type 2 resistance tohead blight infection in fields., 2016, 66: 350–357.
[10] Jia H Y, Zhou J Y, Xue S L, Li G Q, Yan H S, Ran C F, Zhang Y D, Shi J X, Jia L, Wang X, Luo J, Ma Z Q. A journey to understand wheathead blight resistance in the Chinese wheat landrace Wangshuibai., 2018, 48–59.
[11] Arruda M P, Brown P, Brown-Guedira G, Krill A M, Thurber C, Merrill K R, Foresman B J, Kolb F L. Genome-wide association mapping ofhead blight resistance in wheat using Genotyping-by-sequencing., 2016,9. https://doi: 10.3835/plantgenome2015.04.0028.
[12] Rasheed A, Wen W E, Gao F M, Zhai S N, Jin H, Liu J D, Guo Q, Zhang Y J, Dreisigacker S, Xia X C, He Z H. Development and validation of KASP assays for genes underpinning key economic traits in bread wheat., 2016, 129: 1843–1860.
[13] 张宏军, 宿振起, 柏贵华, 张旭, 马鸿翔, 李腾, 邓云, 买春艳, 于立强, 刘宏伟, 杨丽, 李洪杰, 周阳. 利用基因功能标记选择提高黄淮冬麦区小麦品种对赤霉病的抗性. 作物学报, 2018, 44: 505–511. Zhang H J, Su Z Q, Bai G H, Zhang X, Ma H X, Li T, Deng Y, Mai C Y, Yu L Q, Liu H W, Yang L, Li H J, Zhou Y. Improvement of resistance of wheat cultivars tohead blight in the Yellow-Huai rivers valley winter wheat zone with functional marker selection ofgene., 2018, 44: 505–511 (in Chinese with English abstract).
[14] Yu J B, Bai G H, Cai S B, Ban T. Marker-assisted characterization of Asian wheat lines for resistance tohead blight., 2006, 113: 308–320
[15] Stacey J, Isaac P G. Isolation of DNA from plants., 1994, 28: 9–15.
[16] Cui F, Zhang N, Fan Xiao, Zhang W, Zhao C, Yang L, Pan R, Chen M, Han J, Zhao X, Ji J, Tong Y, Zhang H, Jia J, Zhao G, Li J. Utilization of a wheat 660K SNP array-derived high-density genetic map for high-resolution mapping of a major QTL for kernel number., 2017, https://doi:10.1038/s41598-017-04028-6.
[17] Stam P. Construction of integrated genetic linkage maps by means of a new computer package: JoinMap., 1993, 3: 739–744.
[18] 王建康. 数量性状基因的完备区间作图方法. 作物学报, 2009, 35: 239–245. Wang J K. Inclusive composite interval mapping of quantitative trait genes., 2009, 35: 239–245 (in Chinese with English abstract).
[19] Zhai S N, He Z H, Wen W E, Jin H, Liu J D, Zhang Y, Liu Z Y, Xia X C. Genome-wide linkage mapping of flour color-related traits and polyphenol oxidase activity in common wheat., 2016, 129: 377–394.
[20] 李慧慧, 张鲁燕, 王建康. 数量性状基因定位研究中若干常见问题的分析与解答. 作物学报, 2010, 36: 918–931. Li H H, Zhang L Y, Wang J K. Analysis and answers to frequently asked questions in quantitative trait locus mapping., 2010, 36: 918–931 (in Chinese with English abstract).
[21] Srinivasachary, Gosman N, Steed A, Simmonds J, Leverington- Waite M, Wang Y, Snape J, Nicholson P. Susceptibility tohead blight is associated with thesemi-dwarfing allele in wheat., 2008, 116: 1145–1153.
[22] Srinivasachary, Gosman N, Steed A, Hollins T, Bayles R, Jennings P, Nicholson P. Semi-dwarfingandloci of wheat differ significantly in their influence on resistance tohead blight., 2008, 118: 695–702.
[23] Buerstmaye H, Ban T, Anderson J A. QTL mapping and marker-assisted selection forhead blight resistance in wheat: a review., 2009, 128: 1–26.
[24] Jaiswal V, Gahlaut V, Mathur S, Agarwal P, Khandelwal M K, Khurana J P, Tyagi A K, Balyan H S, Gupta P K. Identification of novel SNP in promoter sequence ofassociated with grain weight and other agronomic traits in wheat (L.)., 2015, 10: e0129400.
[25] Somers D J, Fedak G, Savard M. Molecular mapping of novel genes controllinghead blight resistance and deoxynivalenol accumulation in spring wheat., 2003, 46: 555–564.
[26] 朱展望, 徐登安, 程顺和, 高春保, 夏先春, 郝元峰, 何中虎. 中国小麦品种抗赤霉病基因的鉴定与溯源. 作物学报, 2018, 44: 473–482. Zhu Z W, Xu D A, Cheng S H, Gao C B, Xia X C, Hao Y F, He Z H. Characterization of Fusarium head blight resistance geneand its putative ancestor in Chinese wheat germplasm., 2018, 44: 473–482 (in Chinese with English abstract).
[27] 程顺和, 张勇, 张伯桥, 高德荣, 吴宏亚, 陆成彬, 吕国锋, 王朝顺. 小麦抗赤霉病育种2条技术路线的探讨. 扬州大学学报(农业与生命科学版), 2003, 24(1): 59–62. Cheng S H, Zhang Y, Zhang B Q, Gao D R, Wu H Y, Lu C B, Lyu G F, Wang C S. Discussion of two ways of breeding scab resistance in wheat.(Agric Life Sci Edn), 2003, 24(1): 59–62 (in Chinese with English abstract).
Mapping and genetic analysis of QTLs forhead blight resistance to disease spread in Yangmai 16
HU Wen-Jing1, ZHANG Yong1, LU Cheng-Bin1, WANG Feng-Ju2, LIU Jin-Dong2, JIANG Zheng-Ning1, WANG Jin-Ping2, ZHU Zhan-Wang2, XU Xiao-Ting2, HAO Yuan-Feng2, HE Zhong-Hu2,3, and GAO De-Rong1,*
1Lixiahe Institute of Agriculture Sciences / Key Laboratory of Wheat Biology and Genetic Improvement for Low & Middle Yangtze Valley, Ministry of Agriculture and Rural Affairs, Yangzhou 225007, Jiangsu, China;2National Wheat Improvement Center / Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China;3CIMMYT-China Office, Chinese Academy of Agricultural Sciences, Beijing 100081, China
head blight (FHB) resistance of Yangmai wheat cultivars has been paid much attention, but the underlying genetic mechanism is unclear. In recent years, Yangmai 16 is a predominant wheat cultivar durably resistant to FHB in production. A population of 174 double haploid lines (DH) produced by crossing Yangmai 16 (YM16) with the susceptible cultivar Zhongmai 895 (ZM895) was evaluated for FHB response using point inoculation from 2017 to 2019. The DH population was genotyped with wheat 660K SNP array and a high-density genetic map was constructed. Six resistance QTLs were detected, and among them, five were from the resistant parent Yangmai 16 and one from Zhongmai 895.andwere detected at least in two years, explaining 8.8% to 15.0% of the phenotypic variances, respectively.andwere detected only in one year, accounting for 10.5% and 14.7% of the phenotypic variances.andwere detected in one year, too, accounting for 6.4% and 8.3% of the phenotypic variances, respectively. Pyramiding ofmultiple resistant loci with large effects (>10%) is an effective approach to increase FHB resistance. The QTLs identified from Yangmai 16 in the present study will provide a starting point for genetic studies of other Yangmai cultivars, and the QTLs closely linked to markers will be useful for marker-assisted selection in wheat FHB improvement.
;head blight; QTL; marker-assisted breeding
10.3724/SP.J.1006.2020.91048
本研究由国家自然科学基金项目(31901544), 国家现代农业产业技术体系建设专项(CARS-03-03B, CARS-3-2-11), 国家重点研发计划项目(2017YFD0100801, 2017YFD0101802)和江苏省自然科学基金项目(BK20171279)资助。
This study was supported by the National Natural Science Foundation of China, China Agriculture Research System (CARS-03-03B, CARS-3-2-11), the National Key Research and Development Program of China, and the Natural Science Foundation of Jiangsu Province (BK20171279).
高德荣, E-mail: gdr@wheat.org.cn
E-mail: huren2008@126.com
2019-07-22;
2019-09-26;
2019-10-09.
URL: http://kns.cnki.net/kcms/detail/11.1809.s.20191008.1730.006.html
