一对大前庭导水管综合征育龄夫妇遗传咨询研究
2019-12-25唐克峰沈国松娄志武
唐克峰 沈国松 李 志 娄志武
大前庭导水管综合征(larged vestibular aqueduct syndrome,LVAS)是一种以前庭水管扩大伴有感音神经性聋为特征的常染色体隐性遗传性非综合征型听力障碍疾病,其发病与SLC26A4 基因密切相关[1]。SLC26A4 基因位于7q31,含有21 个外显子,开放阅读框架2343bp,每个内含子与外显子的界线分明,SLC26A4 基因编码一个结构和功能都十分复杂的蛋白质Pendrin[2]。该蛋白由780 个氨基酸组成,主要表达于内耳的内淋巴管和淋巴囊,介导OH-、I-、Cl-和HCO3-转运,维持内淋巴液的离子平衡,对内耳内淋巴液再吸收具有不可或缺的作用[3]。SLC26A4 基因的突变谱很广泛,目前世界范围内约有240 种以上的突变报道与耳聋相关[4]。本研究中,笔者对1 对大前庭导水管综合征听障育龄夫妇应用目标区域捕获测序技术对听力障碍相关基因进行突变检测,明确其突变类型和突变形式,并探讨其遗传风险。
1 资料与方法
1.1 研究对象 丈夫30 岁,妻子29 岁,夫妻自诉出生时无异常,无耳毒性药物应用史,无外伤史,皆为2岁前出现对声音反应差,男方不能言语至今,女方可发出少量分辨不清晰言语。听力学检测皆示双耳重度感音神经性聋,夫妻双方MRI 检查结果皆显示双侧内淋巴囊扩大,提示大前庭导水管综合征(见图1)。专科检查:夫妻双方双侧耳廓未见畸形,外耳道及鼓膜未见明显异常。男方父母及姐姐为聋哑人,但均拒绝基因检测;女方为领养儿,家系成员情况不详。另外选取100 名在我院做耳聋基因筛查听力正常且无听力障碍家族史的儿童作为正常对照组,均进行SLC26A4 基因60 个突变位点的检测。本研究经医院伦理委员会审核通过(批号2013-007),研究对象均已签署知情同意书。
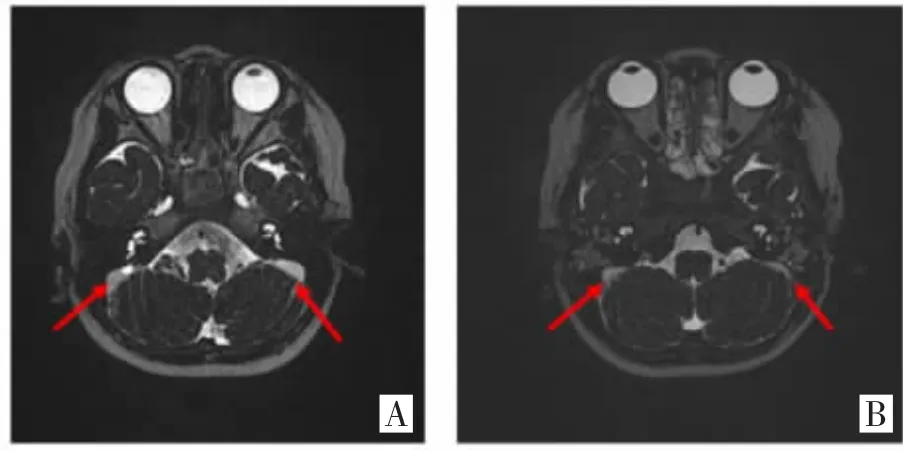
图1 大前庭导水管颞骨MRI 示意图(A:男性;B:女性)
1.2 方 法
1.2.1 DNA 提取 应用DNA 提取试剂盒(厦门致善生物公司,货号603003,批号190614)提取所有耳聋患者的基因组DNA,利用紫外分光光度计测量待检DNA 浓度及纯度,取一定量的DNA 原液稀释成20ng/μL 的工作浓度,-20℃保存备用,DNA 原液-70℃保存。
1.2.2 文库构建及测序 使用Aligent SureSelect 方法制备检测样本,实验方法遵照操作手册,实验包括DNA 片段化处理、末端修复、加接头、PCR 扩增、探针杂交、磁珠捕获富集等步骤。文库经2200 生物分析仪(Agilent)质控分析合格后上机测序。采用Illumina HiSeq X-Ten 系统完成高通量测序。
1.2.3 生物信息学分析 利用FastQC 软件对原始测序数据进行质控分析;利用BWA 软件(v0.7.15-r1140)将所有过滤后的测序序列(reads)比对到参考基因组(GRCh37/hg19);利用Picard 软件工具去除重复reads;利用GATK 软件工具包(v3.7-0)完成单碱基变异(SNV)和插入缺失变异(Indel)的检出;利用Annovar 及VEP 等软件包进行注释。
1.2.4 致病性变异过滤及筛选 结合人群基因变异数据库[dbSNP 数据库(SNP150)、千人基因组数据库等]信息,去除MAF>0.01(AD)或MAF>0.05(AR)的高频变异。参考美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)相关指南,结合疾病基因变异数据库(Clinvar、HGMD 数据库、OMIM 数据库)信息、文献报道、功能试验、遗传模式、基因型-表型关联分析、预测软件(mcap、REVEL 及CADD 等)预测结果等综合判断,将变异分成5 类:致病、可能致病、意义不明确、可能良性、良性;并筛选出与先证者表型相关的可疑致病性变异进行Sanger 测序验证等进一步分析。
1.2.5 Sanger 测序验证及共分离分析 通过以上方法筛选出的可疑致病基因变异,利用Primer3 软件设计PCR 引物进行扩增(PCR 引物由中翰金诺医学研究所设计,华大基因合成,见表1)及Sanger 测序分析。利用Mutation Surveyor 等软件对测序数据进行分析。
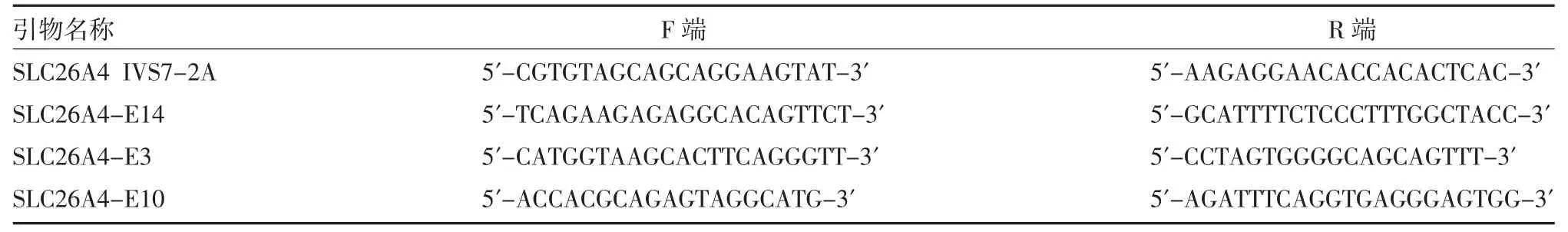
表1 PCR 引物序列
2 结果
本研究发现男方为SLC26A4c.230A >T、c.1174A>T 和c.1547dupC 的复合杂合突变,Sanger 测序示意图分别如图2 所示。女方为SLC26A4c.919-2A>G 和c.1547dupC 的复合杂合突变,Sanger测序示意图分别如图3 所示。而正常对照组100名检测对象里SLC26A4 基因60 个位点未见任何突变。
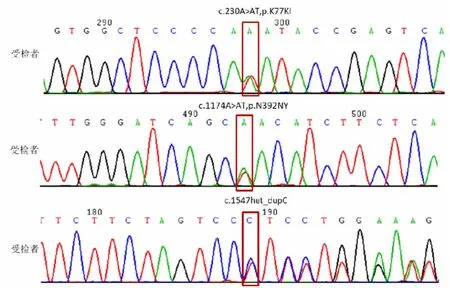
图2 本例聋哑夫妇男性的SLC26A4 基因突变位点Sanger测序结果
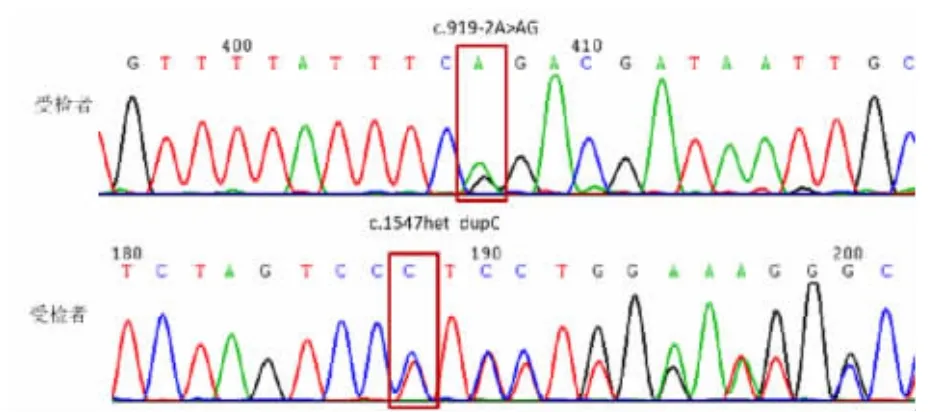
图3 本例聋哑夫妇女性的SLC26A4 基因突变位点Sanger 测序结果
3 讨论
20 世纪70 年代末随着CT 问世,LVAS 逐渐被认识,它是遗传性耳聋患儿中最常见的一种内耳发育畸形性疾病,也是一种常染色体隐性遗传性听力障碍性疾病[5]。其主要表现为双侧进行性感音神经性耳聋,影像学CT 或MRI 提示内淋巴囊或前庭导水管扩大[6]。患病个体间听力损伤程度差别较大,听力范围从完全正常到极重度听力障碍不等,咳嗽、头部倒立、用力擤鼻涕以及剧烈体育运动等都可以造成听力损伤。早期临床诊断或者基因诊断,结合正确的防护措施,有助于保护患者的听力,甚至能够完全正常生存,提高生活质量。
中国耳聋高发与中国人群常见耳聋基因突变的高携带率息息相关,其中SLC26A4 基因突变的携带率高达2%~3%,约80%LVAS 患者中可见SLC26A4突变[7]。再加上因为社会原因,耳聋患者结为夫妇的概率大大增加。由于其耳聋基因携带率高,遗传性耳聋比例高,从而导致生育后代耳聋概率大大高于普通婚配夫妇,因此耳聋夫妇成为高危人群应该受到重点预防和干预。
有研究报道,SLC26A4 基因在我国最常见突变位点是c.919-2A>G、c.2168A>G、c.1174A>T、IVSl5+5G>A、c.1229C>T 及c.1975G>C,并且具有种族特异性[3]。其中c.919-2A>G 单等位基因突变频率为7.6%,该突变占SLC26A4 外显子7 和8 中所有突变等位基因的99%,但复合杂合性在两个突变等位基因的患者中占近60%,由此可见针对SLC26A4 基因所有位点进行筛查是很有必要的[8-10]。c.919-2A>G 为剪切位点变异,属于功能缺失型变异。该变异可导致Pendrin 蛋白发生功能障碍,使得内耳内的淋巴液流动发生异常,从而引起内淋巴管内压升高和前庭导水管扩大,最终内耳毛细胞受损和听神经萎缩出现听力下降[5]。c.1174A>T 变异较c.919-2A>G 罕见,属于错义突变,导致氨基酸N392Y 改变,肖彩霞等[11]报道了1 例,李琦等[12]报道了3 例,Liu 等[13]报道过2例,但都没有阐述具体的致病机制。C.230A>T 和c.1547dupC 更是罕见,其中c.230A>T 属于错义变异,而c.1547dupC 属于框移变异。C.230A>T 突变导致K771 氨基酸的改变,从而影响整个pendrin 蛋白的功能[14]。C.1547dupC 突变导致了S517fs 氨基酸的改变,Liu 等[13]也报道过2 例。C.230A>T、c.1174A>T 和c.919-2A>G 虽然都是罕见的突变,但是这三种突变极少出现纯合突变,基本上都是以和SLC26A4 基因其他突变位点复合杂合的形式出现,都会导致不同程度的耳聋。
本研究中男性患者属于SLC26A4 基因三个致病杂合突变的复合杂合子,而女性患者在相同的基因两个致病杂合突变的复合杂合子,其中一个突变与男性相同。该男性的父母及姐姐都是聋哑人,很可能是从其父母SLC26A4 基因不同突变位点遗传得来,导致三个位点的复合杂合子,这是比较罕见的,遗憾的是他的父母及姐姐拒绝基因检测。而女方是比较常见的LVAS 复合杂合子患者,其基因的来源也因为是领养儿而不能追寻。但根据单基因遗传病的孟德尔遗传定律,该对夫妇生育LVAS 患儿的风险为100%,可不生育、领养、供精或供卵形式获取健康后代。由此可见,我们通过对聋人夫妇的耳聋基因进行检测,并进行遗传咨询,可以避免遗传风险明确的夫妇生育耳聋后代,有效降低耳聋的再发风险。
