基于5-硝基间苯二甲酸配体的六核Co(II)配合物的合成、结构及磁性
2019-12-23赵振新张浩浩何坤欢章应辉
赵振新,程 琪,张浩浩,何坤欢,章应辉
(1.河南城建学院材料与化工学院,平顶山 467036;2.南开大学材料科学与工程学院,天津 300350)
1 引 言
近二十年来,配合物因其纷繁多样的拓扑结构和物化性质引起了科研工作者的广泛关注,并在气体存储和分离[1-4]、多相催化[5-6]、分子磁体[7-8]、光学[9]、分子识别和化学传感[10-11]等领域显示出诱人的应用前景。其中,Co(II)离子在八面体配位环境下具有高自旋电子结构,其簇合物在一定条件下展现出有趣的磁现象,在高密度信息存储和量子计算领域有重要的潜在应用,成为分子磁学工作者们的一个研究热点[12-18]。查阅大量文献发现,通过合理设计和精确挑选短桥有机配体能够在一定程度上实现分子基磁性Co(II)配合物的可控合成[19-23]。
最近,人们基于混合配体构筑策略报道了系列新颖和功能优异的离散簇合物和配位聚合物[24-28]。通过合理的混合配体选择可以在金属簇间实现较强相互连接,是构筑新型分子基磁性配合物的有效方法。本文选择5-硝基间苯二甲酸(H2nip)和线形柔性的4,4′-二吡啶胺配体(dpa)来构筑新型Co(II)基配合物。H2nip具有一定的刚性,其两个羧基可以提供更多的配位点来构筑双核和多核簇基元。同时,羧基还可以有效传递磁性,有利于配合物磁学性质的表现。线形柔性的dpa具有小的空间位阻,容易取代金属簇上的配位溶剂分子,在金属簇间形成有效联接,进而形成高维复杂的网络拓扑结构。
本文采用5-硝基间苯二甲酸和4,4′-二吡啶胺混配策略,在不同溶剂热反应条件下得到了两例钴(Ⅱ)配合物1和2。晶体结构解析表明配合物1和2有相似的六核簇结构单元,但是显示不同的拓扑网络,表明溶剂对晶体结构设计有重要的影响。磁学性质测试二者均呈现反铁磁相互作用。
2 实 验
2.1 试剂和仪器
除4,4′-二吡啶胺配体根据文献[29]合成外,其他所用化学试剂均为市售商品化试剂。所用溶剂均为国产分析纯,未经任何处理直接使用。元素C、H、N含量分析测定使用Perkin-Elmer 240型元素分析仪;红外光谱在TENSOR27 OPUS(BRIIXER)傅立叶变换红外光谱仪上测得;荧光光谱由FL-4500荧光光谱仪测定;磁性质测试由QuantumDesignMPMSXL-7 SQUID磁力计测定;X-射线粉末衍射(XRD)由RigakuD/Max-2500X-射线粉末衍射仪测定。
2.2 配合物1、2的合成
{[Co3(μ3-OH)(μ2-OH)(nip)2(dpa)(MeOH)(H2O)]·2H2O}n(1):将Co(NO3)2·6H2O (145.5 mg, 0.5 mmol),NaOH (40 mg, 1.0 mmol),H2nip (106 mg, 0.5 mmol),4,4′-dipyridylamine (85 mg, 0.5 mmol) 和15 mL溶剂(V水∶V甲醇=2∶1)混合,充分搅拌后把悬浊液转移到聚四氟乙烯高压釜中加热,在12 h内升温至165 ℃,并在165 ℃下恒温4 d,然后以5 ℃/h的速率缓慢冷却至室温,所得产物用 5 mL 乙醇洗涤两次。在空气中干燥后得到浅红色透明多面体柱状晶体。基于金属Co计算产率为27%。元素分析:C27H21Co3N5O18;计算值/%:C, 36.84; H, 2.40; N, 7.95;实验值/%:C, 36.89; H, 2.32; N, 7.85。IR (KBr, cm-1):3458m, 3146m, 1586s, 1524s, 1382s, 1339s, 1213s, 994m, 813m, 725m, 523m。
{[Co3(μ3-OH)2(nip)2(dpa)(EtOH)(H2O)]·2H2O}n(2):合成过程类似配合物1,溶剂采用(V水:V乙醇=2∶1),得到紫红色柱状晶体。基于金属Co计算产率为22%;元素分析:C28H29Co3N5O18。计算值:C, 37.35; H, 3.24; N, 7.77 %;实验值:C, 37.15; H, 3.14; N, 7.97 %。IR (KBr, cm-1):3452m, 3143m, 1581s, 1522s, 1383s, 1336s, 1213s, 997m, 812m, 726m, 524m。
2.3 配合物1、2晶体结构测定
在显微镜下选取合适大小的单晶,室温下在Rigaku SCX-min衍射仪上进行单晶测试,以经石墨单色器单色化的Mo-Kα射线(λ=0.071073 nm)为光源,以ω方式收集衍射数据。衍射数据使用SADABS程序进行半经验吸收校正[30]或使用multiscan程序进行洛伦兹偏振和吸收校正[31]。晶胞参数用最小二乘法确定。
3 结果与讨论
3.1 配合物1、2晶体结构参数
单晶衍射数据还原和结构解析分别使用SAINT[32]和SHELXTL[33]程序完成。所有的晶体结构都用直接法解出,先用差值傅立叶函数法和最小二乘法确定全部非氢原子坐标,并用理论加氢法得到氢原子位置,然后用最小二乘法对晶体结构进行各向异性精修,配合物2中的乙醇分子的5个氢原子没有加上。配合物1和2的主要晶体学参数列于表1中(CCDC:1,1855840;2,1855839)。
3.2 配合物1、2的晶体结构描述
{[Co3(μ3-OH)(μ2-OH)(nip)2(dpa)(MeOH)(H2O)]·2H2O}n
(1)
配合物1结晶于单斜晶系,空间群为P21/n,其不对称基本结构单元包含三个Co中心、两个nip2-、一个4,4′-二吡啶胺﹑一个MeOH﹑两个OH-﹑两个晶格水(图1(a))。Co1与五个氧、一个氮原子(O1、O11、O15、O15A、N3和O13B)配位形成CoO5N八面体构型,其配位原子分别来自两个不同的nip2-配体、一个dpa和三个OH-。Co2与六个氧原子配位形成CoO6八面体构型,配位原子(O5、O9、O13、O11、O15和O16)则分别来自三个不同的nip配体、一个配位水分子和两个OH-。Co3与五个氧、一个氮原子配位形成CoO5N八面体构型,配位原子(O2、O6、O12、O11、N5和O14)分别来自三个不同的nip配体、一个dpa和一个MeOH。配合物1中的Co-O键长在0.200~0.237 nm范围。
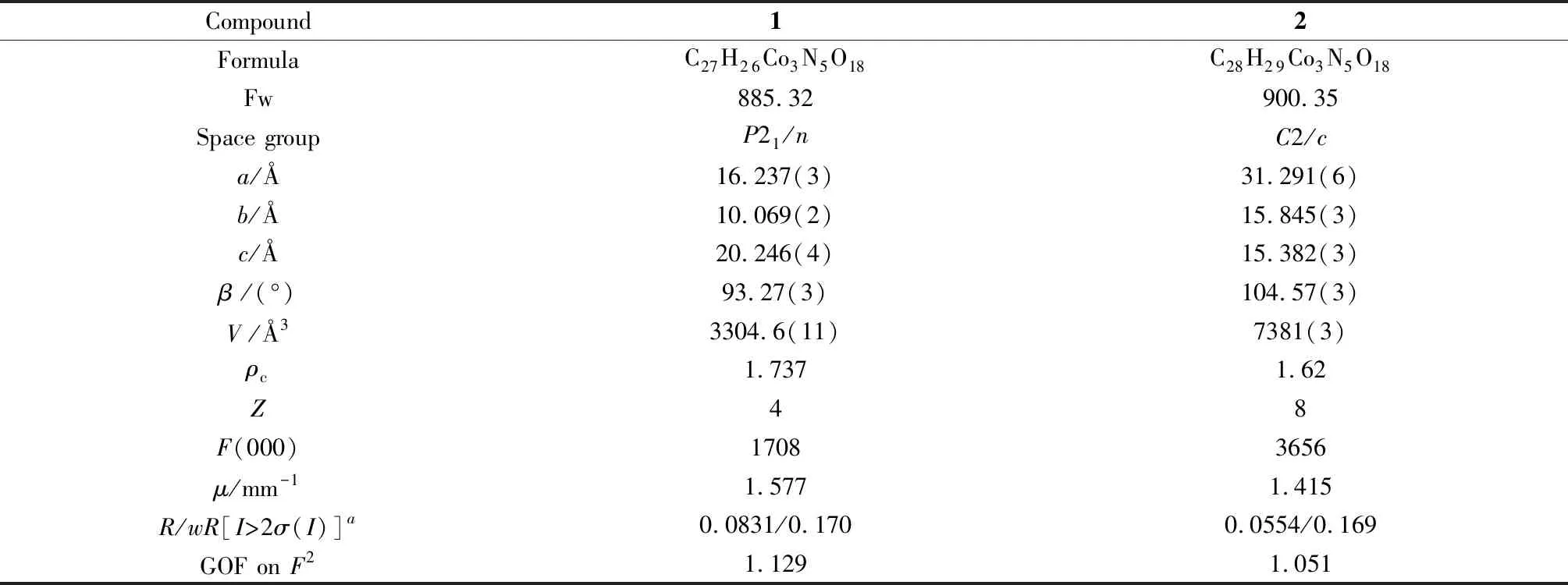
表1 配合物1和2的主要晶体学参数Table 1 Main crystal parameters of compound 1 and 2
图1 (a) 配合物1中CoII离子的配位环境;(b) 六核钴簇的连接模式;(c) 二维层结构图;(d) 三维堆积结构图Fig.1 The crystal structure of compound 1: (a) the coordination situation of Co(II) ion; (b) the linking mode of hexanuclear cluster;(c) the two dimensional layer; (d) the three dimensional packing mode
在配合物1中,六个钴中心(Co1、Co1A、Co2、Co2A、Co3和Co3A)通过四个OH-构成一个六核钴簇[Co6(OH)4(OCO)8]次级结构单元(图1(b))。六核钴簇通过nip2-配体形成一个二维层结构(图1(c)),二维层进一步通过dpa配体连接形成三维配位网络(图1(d))。nip2-和dpa均采用二连接模式桥联相邻两个六核钴簇,而六核钴簇可看做10连接节点。通过这种简化方式,配合物1呈现出单节点10-连接的三维pct 类型拓扑网络结构(图2), Schäfli符号为(312·428·55)。
{[Co3(μ3-OH)2(nip)2(dpa)(EtOH)(H2O)]·2H2O}n
(2)
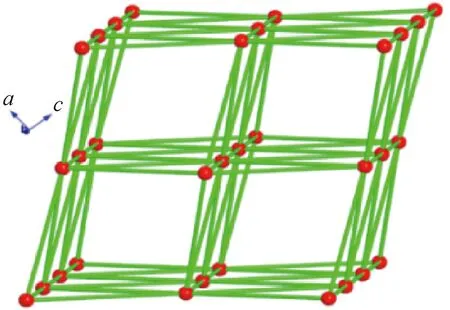
图2 配合物1拓扑网络的结构图Fig.2 The topological structure of compound 1
配合物2属于单斜晶系,空间群为C2/c,其不对称单元包含三个Co中心、两个nip2-﹑一个dpa﹑一个配位水分子﹑一个EtOH分子﹑两个OH-和两个晶格水(图3(a))。Co1和Co2 均以六配位形式存在,处于八面体配位几何构型环境中,Co3则是五配位的变形的三角双锥几何构型。Co1分别与一个nip2-、一个dpa﹑两个OH-﹑一个EtOH分子和一个水分子配位形成CoO5N八面体。Co2分别与两个nip2-、一个EtOH分子和三个OH-配位形成CoO6八面体。Co3与三个nip2-配体、一个dpa和一个OH-配位形成CoO4N畸变三角双锥结构。Co-O键长范围在0.199~0.230 nm内。
图3 配合物2的晶体结构Fig.3 The crystal structure of compound 2
六个相邻钴中心(Co1、Co1A、Co2、Co2A、Co3和Co3A)通过四个OH-和两个μ2-EtOH构成一个六核钴簇[Co6(EtOH)(OH)4(OCO)8]次级结构单元(图3(b))。每个六核钴簇通过nip2-与周围四个相邻六核钴簇相连,形成二维层结构(图3(c))。二维层间通过dpa配体相连接形成三维配位网络(图3(d))。nip2-和dpa配体均可视为2-连接的连接杆桥联相邻两个六核钴簇,而六核钴簇可视为6-连接节点与四个nip2-、两个dpa配体连接。从拓扑观点来看,配合物2可简化为单节点6-连接的三维pcu类型拓扑网络结构(图4),Schäfli符号为(412·63)。比较1与2结构可以发现,1中甲醇分子的氧原子作为单配位点参与了配位,而2中的乙醇分子桥联了两个相邻的Co1和Co2,这导致二者虽然具有相似的六核簇结构单元,但却表现出不同的拓扑网络结构。
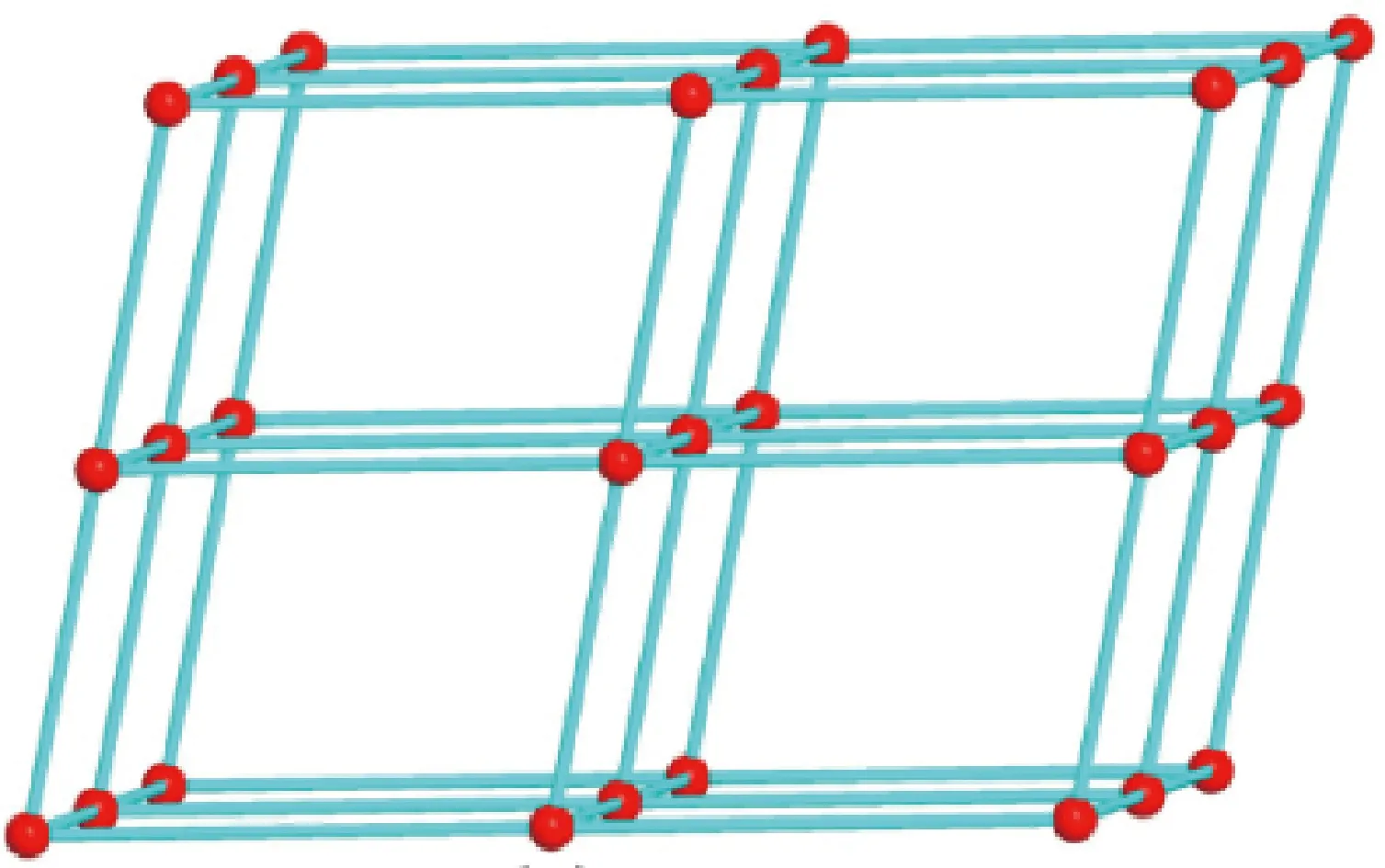
图4 配合物2拓扑结构图Fig.4 The topological structure of compound 2
3.3 粉末XRD分析
图5为配合物1和2的实测和模拟粉末XRD图谱。两个配合物样实测粉末XRD图谱峰位和通过单晶结构数据模拟得到的峰位基本吻合,表明这两个配合物样品具有较高的相纯度。衍射峰强度的差别可能是由晶体在粉末样品中的择优取向所致。
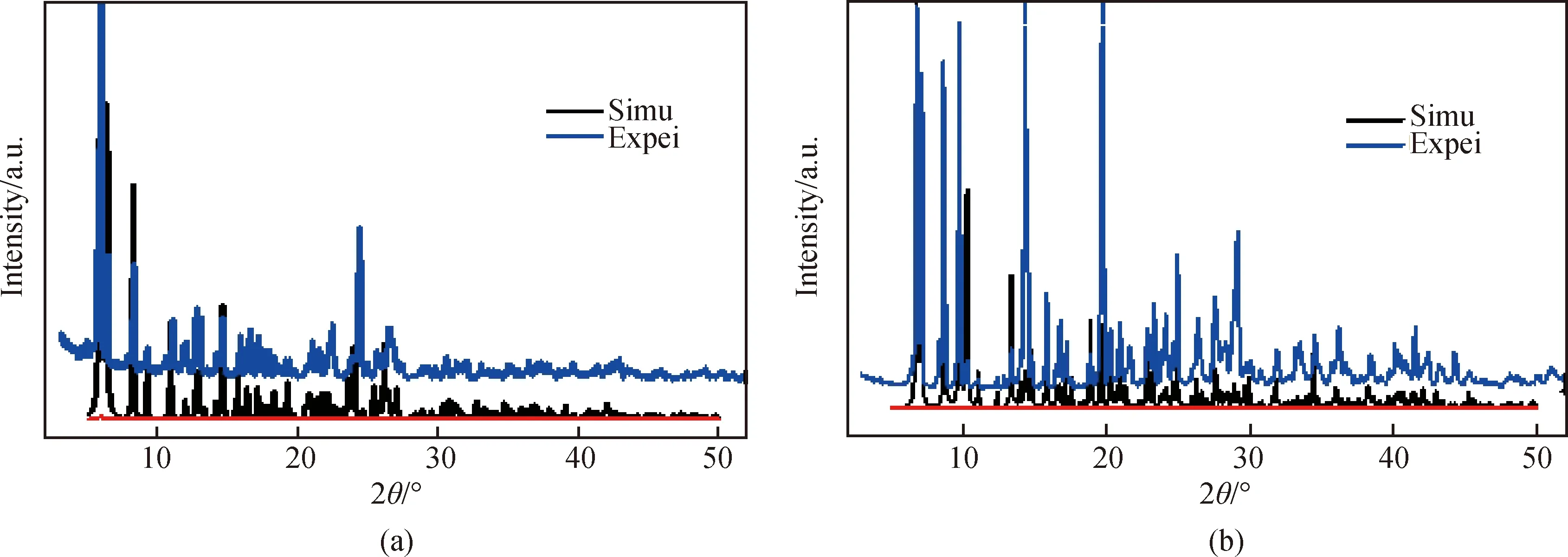
图5 配合物(a)1和(b)2的 XRD图谱Fig.5 XRD patterns of compound (a)1 and (b)2
3.4 磁学性质分析
测量了2~300 K温度范围内配合物1和2的摩尔磁化率(图6)。室温下,其χMT值分别为8.33 emu·mol-1·K和8.24 emu·mol-1·K,远大于g=2.0时三个孤立CoII离子的χMT值(5.625 emu·mol-1·K),这是由钴离子的旋轨耦合导致的。随着温度的降低,2 K时两个配合物的χMT值逐渐降低至1.33 emu·mol-1·K和1.44 emu·mol-1·K。对16~300 K间的数据进行居里外斯拟合(χM=C/(T-θ)),得到1和2的相关参数分别为C=9.48 emu·mol-1·K、9.22 emu·mol-1·K和θ=-39.14 K、-27.64 K。值得注意的是,钴离子旋轨耦合对负的外斯常数是有贡献的,其作用强度可以用公式(1)估算。
χT=Aexp(-E1/kT) +Bexp(-E2/kT)
(1)
公式中,A+B等于居里外斯常数,而E1和E2分别代表相对应反铁磁超交换作用和旋轨耦合的活化能。对2~300 K区间实验数据的最佳拟合给出配合物1和2的相关参数A+B分别为 9.57 emu·mol-1·K、9.06 emu·mol-1·K, -E1/k分别为-1.09 K、-1.22 K,-E2/k分别为-67.70 K、-30.35 K。-E1/k较小的负值说明钴离子间为是弱反铁磁相互作用。
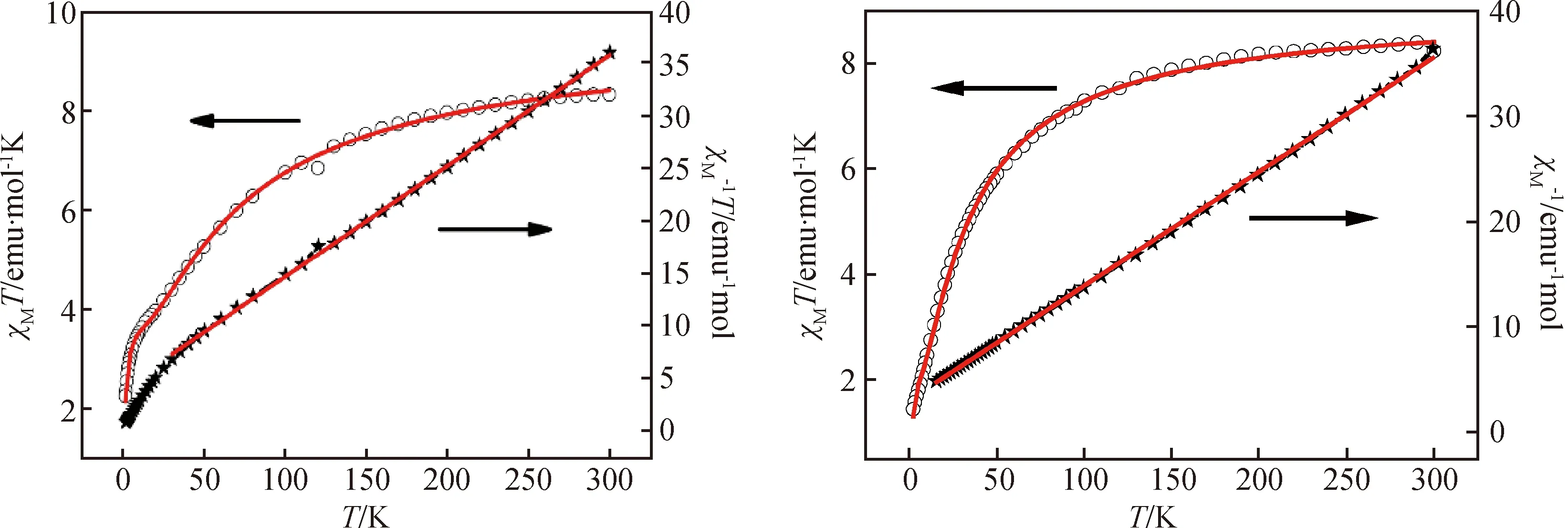
图6 1000 Oe场下配合物1和2的χMT vs.T曲线(红色实线部分为2~300 K的拟合值)和χM-1 vs. T曲线(实线部分为居里外斯拟合值)。Fig.6 The χMT vs. T plots and χM-1 vs. T plots of compound 1 and 2 under 1000 Oe. The solid line is the simulated one
以上分析表明,配合物1和2中钴六核簇内钴离子之间是反铁磁相互作用,六核簇间也呈现反铁磁相互作用的,使得1和2整体上呈现出反铁磁行为。进一步测试了配合物2的Mvs.H曲线(图7)。可以看出,M随着外场的增加缓慢增加,在70 kOe时达到2.70 Nβ。该数值远小于三个CoII的饱和值6.6 Nβ,进一步证明2中钴离子之间是反铁磁相互作用的。另外,配合物1和2均含有μ3-OH和syn-syn羧酸桥联模式。μ3-OH连接对应的Co-O(H)-Co角度在94°~107°范围内,有可能产生铁磁性。这说明syn-syn的羧酸磁交换通道可能在配合物的反铁磁相互作用中起关键作用。
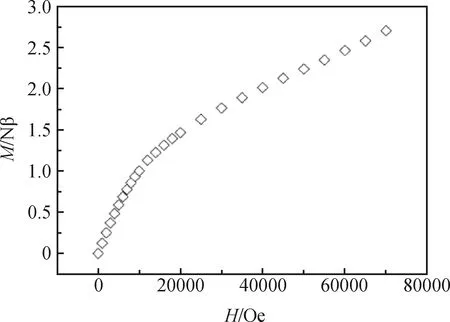
图7 2 K时配合物2的M vs.H曲线Fig.7 The M vs. H plot of compound 2 at 2 K
4 结 论
本文通过刚性羧酸配体H2nip和线性柔性吡啶亚胺配dpa混配,在不同溶剂中反应合成了两个含六核簇结构单元的三维Co(II)配合物。两种配合物中羧酸和配位水分子将六核Co(II)簇连接为二维层结构,而中性二吡啶基配体进一步将二维层连接为三维结构。尤其值得关注的是1合成中甲醇溶剂参与配位,而2合成中的乙醇溶剂没有参与配位,这导致二者虽然具有相似的六核簇结构单元,但却表现出不同的拓扑网络结构,表明溶剂对配位聚合物的拓扑设计有重要影响。磁学性质分析显示两例配合物中的Co2+间呈现反铁磁相互作用。
