全外显子测序在糖尿病中的应用
2019-12-21宗奕岑胡承贾伟平
宗奕岑,胡承,贾伟平
上海交通大学附属第六人民医院,上海市糖尿病研究所,上海市糖尿病重点实验室,上海 200233
DNA测序技术诞生于20世纪70年代,发展至今已40余年。从第一代基于Sanger法的DNA测序技术到第二代高通量测序(又称下一代测序,next generation sequencing, NGS),再到第三代单分子测序,乃至最新的第四代固态纳米测序,测序技术发展迅猛并不断变革,测序通量和精确度不断改善,测试时长不断缩短。目前,第二代短读长测序技术在全球测序市场上仍占据优势位置,而第三代和第四代测序技术近几年在肿瘤、免疫等领域飞速发展。测序技术的每一次变革,都对疾病的病因机制研究和药物研发等产生巨大的推动作用。
随着人类基因组计划的完成,其他物种生物的基因组信息也逐渐清晰。研究已发现不同物种间的基因差别仅为1 %,且主要集中在外显子区域。外显子作为DNA中的重要功能序列,包含了合成蛋白质所需的全部信息,是基因发挥其功能最直接的体现。人类外显子组序列仅占人类整个基因组序列的1 %(约30 Mb、18万个外显子),却涵盖了与个体表型/疾病相关的大部分功能变异,包含了约85%的致病突变[1-2]。全外显子组测序(whole-exome sequencing, WES)是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法,相较于全基因组测序来说,更加简便、高效、经济,且具有更高的覆盖度。在临床诊疗疾病的应用中,WES不仅可以加快确定单基因突变疾病致病基因,从而明确临床诊断,而且对于多基因复杂疾病如糖尿病、肥胖、肿瘤等致病基因的发现和易感基因的定位亦具有巨大推动作用[3-4]。本文将围绕全外显子组测序技术及其在糖尿病分子病因机制、临床预警诊疗等方面的应用进行系统阐述。
1 全外显子组测序技术原理概述
WES通过外显子组的捕获富集、高通量测序和生物信息学分析这三个主要步骤,最终定位潜在致病基因/位点。
1.1 外显子组的捕获富集
外显子区域DNA富集,需要将基因组DNA随机打断成200~300 bp的小片段,进行DNA片段末端修复,5'端加磷酸基团,3'端加PloyA尾,经过PCR扩增后形成完整的文库。随后将这些DNA片段与捕获芯片上的引物杂交,分离出引物-DNA复合物,然后洗掉未杂交的DNA片段,将杂交的DNA片段洗脱,得到富集的目标片段[5-6]。最后将目标片段连接成长链DNA,再次随机打断并在其两端连接上测序接头,用与接头相匹配的序列为引物进行PCR扩增,经质量检测合格后的外显子组文库即可上机测序(图1)。目前用于外显子组捕获的平台主要有Agilent公司的SureSelect Human All Exon 50mb、Illumina公司的TruSeq Exome Enrichment、Roche/NimbleGen公司的SeqCap EZ Exome Library2.0。2011年,美国斯坦福大学医学院对这三个平台进行了综合评估,认为:从检出变异体的数量角度来看,NimbleGen平台相较于其他两种平台覆盖了较少的基因组区域,但通过较少测序可以灵敏地检出小变异,而另外两者可以捕获更多变异体;从捕获区域方面来看,NimbleGen探针覆盖到多次捕获的碱基上是三者中密度最高的平台,Agilent探针以紧密相连的方式覆盖整个外显子区,而Illumina基于paried-endreads,将覆盖区域扩大到探针序列以外,且覆盖这些间隔区域,因此能捕获非翻译区序列;从测序深度方面分析,NimbleGen可以富集到更多的目标碱基,Illumina和Agilent在更多的reads数量情况下可富集更多的目标碱基。也就是说:高密度探针设计捕获较少的目标碱基,有更高的变异检出效率;而低密度探针可捕获更多的碱基,但需要更深度的测序。
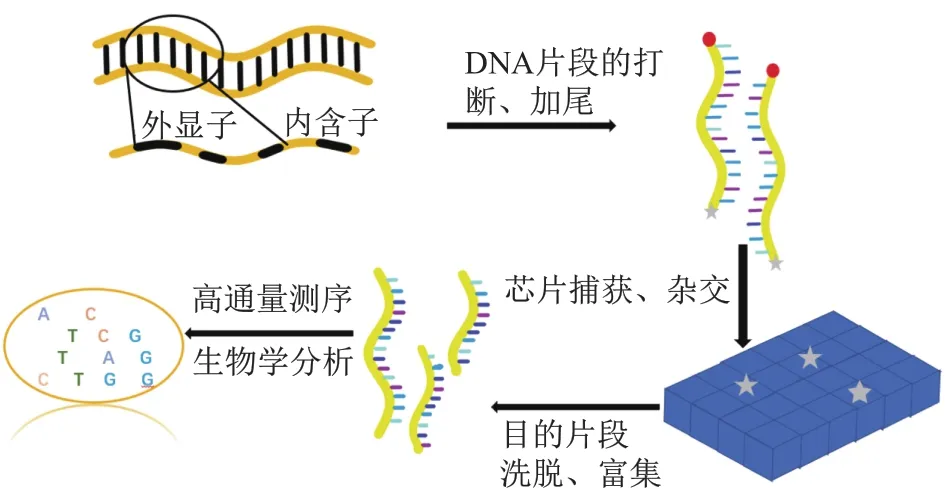
图1 WES流程简图
1.2 高通量测序
高通量测序技术原理是边合成边测序,将片段化的基因组DNA两侧接上通用的测序接头,形成众多PCR单克隆阵列并进行独立的PCR反应,用不同颜色的荧光标记4种不同的dNTP,反应时每添加一种dNTP就会释放不同荧光,捕获并检测荧光信号获得待测DNA序列[7]。随着测序技术的发展,第三代单分子测序仪不需要构建DNA文库,而是基于纳米孔的单分子实时测序技术,测序通量高,测序读长较长,更有利于基因组的拼接,但缺点是错误率较高,需要加大测序深度确保更好的测序精度。目前高通量测序公司主要有Roche 454焦磷酸测序、Illumina Solexa 合成测序及ABI SOLiD连接法测序。相较而言,Illumina Solexa合成测序通量更高,样本量更少,精确性更好,且成本更低,因此适用更加广泛[8]。
1.3 生物信息学分析
WES数据的分析具有一定的挑战性,需要进行大规模的数据处理,很多分析过程及文件的处理只能使用高性能的计算机和编程语言来实现。分析流程主要包括数据质量评估、变异筛选、功能预测、对结果的解释,以及用适当的形式呈现。具体步骤为:测序后首先进行碱基识别(Fastq格式),通过去除接头序列,低质量、长度太短的reads等一系列的质量控制,将过滤后的数据与参考序列(Fasta格式)进行比对,通常需要指定测序靶标区域;得到组装后SAM/BAM格式文件后,再以BAM文件为基础,鉴定核苷酸变异(VCF格式)信息。WES目前已经广泛应用于检测遗传变异,为研究疾病的遗传关联、人类的多样性和种群历史提供了重要的线索。然而,所有这些研究都依赖于对真阳性变异的准确检测和对测序错误、低等位基因分数或较低的测序深度导致的假阳性变异的有效排除。GATK(Genome Analysis Toolkit)是目前检测基因变异最常用的软件之一,对于数据质量的保证高度重视,功能强大且适用于各种规模的数据。通常,得到的原始变异结果中包含有大量的假阳性变异,过滤方法主要有两种:一种是设置参数直接过滤,另一种是使用 GATK的VQSR(突变质量重矫正)。使用VQSR的前提是数据量至少为 30个WES数据,GATK建议使用VQSR。然后利用公共数据库(dbSNP150、1000 genome、ExAc、ESP6500等)对过滤后的突变进行注释,得到突变频率信息。通过去除人群中常见的变异位点,再结合一些功能注释软件,如Polyphen2、SIFT、M-CAP及CADD分数等对过滤后的变异进行蛋白功能影响预测,筛选出候选基因,通过分子生物学和功能实验验证,最终找到致病基因。
2 WES在糖尿病精准诊疗中的应用
WES基于其高特异性、高准确性和高覆盖度的优点,不仅广泛应用于孟德尔遗传病分子病因的解析,在复杂疾病如糖尿病、肥胖和癌症等疾病易感基因的识别方面亦发挥了巨大作用。无论是单基因疾病致病基因的定位,还是多基因复杂并易感基因的识别,都需要检测大量的基因变异,一代测序无疑是研究的瓶颈,而WES则通过提高测序深度就能够准确大量快速地获得所需要的数据。
2.1 单基因突变糖尿病诊疗中的应用
大多单基因突变糖尿病,如青少年发病的成人型糖尿病(MODY)、新生儿糖尿病(NDM)都符合孟德尔遗传定律。由于明确的基因型/表型相关性,解析孟德尔遗传病的分子病因成为理解特定基因如何导致特定疾病及特殊表型的宝贵工具。孟德尔糖尿病的遗传学研究在识别与发病机制有关的基因方面取得了显著成功,定位了包括影响胰岛素分泌(ABCC8、GCK、INS、KCNJ11)及胰腺发育的重要转录因子(HNF1A、HNF4A、PDX1、PTF1A)。随着基因测序技术的不断发展,越来越多的符合孟德尔遗传方式的单基因糖尿病的分子病因被准确解析,如Bonnefond等[9]通过对糖尿病家系的全外显子组分析,发现KCNJ11基因突变Glu227Lys在家系中与糖尿病共分离,由此确认了KCNJ11是该糖尿病家系致病基因,并定位了新的MODY亚型MODY13。此外,De Franco等人对一组胰腺发育不全的患者的先证者进行WES,定位到GATA6基因上的新突变,随后扩大样本量在更多的胰腺发育不全的患者中进行GATA6基因区域靶向测序,发现了该基因区域更多的非同义突变,随访研究证实GATA6基因突变携带者均患有不同程度的胰腺发育障碍和成年发病的糖尿病,但并不伴随外分泌腺功能障碍[10-11]。该研究通过WES不仅证实了GATA6突变是胰腺发育不全的致病原因,还提示了该基因在胰腺发育中的重要作用。对于单基因糖尿病分子病因的解析不仅可以明确患者诊断,还有助于实现个体化治疗。例如,由编码KATP通道亚基的基因(如KCNJ11、ABCC8)上发生的突变而导致的糖尿病,这部分患者可直接应用作用靶点在KATP通道的磺脲类药物精准控制血糖,而不需要几药联合或胰岛素治疗[12]。
2.2 WES在T2D中的应用
近10年,基于“常见变异-常见疾病”的理论模型,国际上通过全基因组关联研究(GWAS)已经发现了100余个2型糖尿病(type 2 diabetes, T2D)易感基因,但集结所有遗传变异的效应,也只能解释T2D 10%~15%的遗传度,也就是说,仍有很多易感基因/变异未被发现,用于解释所谓丢失的遗传度(missing heritability)。尽管高密度SNP芯片也可以检测一些低频和罕见的变异,但芯片的固定位点仅限于检测已知序列的变异。高通量测序却可以发现新的疾病的易感基因或者外显子区域可以导致复杂形状的编码变异。一项研究通过对糖尿病易感基因KCNJ11和HHEX的重新测序发现了在该基因区域内的罕见变异对糖尿病的发生贡献度较高[13],由此,有研究者们提出了“罕见变异-常见疾病”的模型假设。但罕见变异对于复杂疾病T2D的贡献度到底有多大目前仍具争议。Albrechtsen等[14]基于WES技术对1 000例具有代谢异常的患者(包含T2D、肥胖和高血压)和1 000例正常对照人群进行测序,发现COBLL1和MACF1基因常见变异与T2D显著相关。Lohmueller等[15]在2 000例丹麦糖尿病病例对照人群中进行WES,但并未发现与T2D显著相关的罕见变异。这两项研究表明,在一般人群中,少数基因的罕见编码变异在T2D的遗传基础中似乎并未起到主要作用。研究者们推测当聚集在少数基因中时,中到强效应的罕见编码变异不太可能解释大部分缺失的遗传性。如果罕见的编码变异是T2D风险的一个重要因素,它们很可能分散在许多基因上。但是,来自于糖尿病遗传高风险种族的数据却比较令人振奋。一项对Pima印第安人的WES研究发现(N=177),CYB5A(编码酯酰辅酶A脱氢酶)和RNF10基因(功能不明)与美国印第安人群肥胖和T2D风险显著相关[16]。另一项来自于SIGMA type 2 consortium的大型国际合作研究对来自于墨西哥和美国拉丁裔的3 756例受试者开展WES(1794 例T2D,1 962例正常对照),发现胰腺转录因子肝细胞核因子1A(HNF1A)基因与T2D显著相关,其编码区变异p.E508K可增加T2D发生风险5倍以上。该变异在糖尿病患者中频率为2.1%,而在正常人群中仅为0.36%。有趣的是,随后的多种族验证发现该变异仅存在于拉丁裔人群中,进一步机制研究亦证实突变体导致HNF-1A下游靶基因转录活性下降,从而导致糖代谢异常[17]。这两项研究是应用WES解析T2D这种复杂疾病分子病因的初步尝试,尽管存在一些局限,但仍为定位T2D易感基因提供了新的方向。最近,全球百余名遗传学者联合在《自然》杂志公布了目前全球最大的T2D的全外显子组测序结果(涉及约4.5万病例/对照人群:20 791例2型糖尿病患者及24 440例正常糖调节人群),研究者希望能够通过全外显子组的大数据发现影响疾病风险的编码蛋白质的遗传变异,为疾病发病机制提供相关线索。研究者对来自5个不同种族(西班牙裔、欧裔、非裔美国、东亚及南亚人群)的4万余例人群开展全外显子组测序,发现MC4R、PAM、SLC30A8和UBE2NL这4个基因与T2D显著相关,其中UBE2NL基因是新发现的T2D相关基因。随后研究者还比较了WES与GWAS对T2D的解析度,发现从GWAS中选取的与T2D最为相关的前100个基因信号在本研究人群中仅解释了1.96%的遗传变异度,而由WES发现的T2D最强罕见变异信号,可解释25%的由GWAS发现的最强常见变异导致T2D的遗传度[18]。
3 展望
随着测序通量的不断加深及技术的不断普及,基因组范围的测序不仅对于认识糖尿病的病理生理机制具有重要意义,还有利于糖尿病的精准诊断和个体化治疗,并有望发现新的药物靶点。未来通过GWAS、二代甚至是三代测序可能会发现成百上千个糖尿病相关变异。对于性状复杂的T2D来说,GWAS发现的常见变异与WES的罕见变异对于揭示T2D的遗传特点仍具有互补性,前者有利于新基因新位点的发现,而后者则侧重于挖掘新基因新位点的潜在信息。此外,对于临床医生所关注的T2D精准医疗来说,外显子测序可以产生一些具有足够效应的罕见变异,并对T2D遗传风险预测提供一定贡献度。然而,在目前已知的T2D药物靶点基因中发现的罕见变异,其对于疾病的效应需要样本量高达75 000~185 000个测序方可达到全外显子组的统计学效力,因此,在实际人群中,由外显子组测序发现的罕见变异需要和由GWAS所产生的多基因位点评分共同参与评估疾病的风险和个体化治疗效果。
