碱水解-SPE-LC-MS/MS法快速测定动物源食品中喹乙醇代谢物残留量
2019-12-19余婷婷
刘 迪 韩 莉 曾 妮 余婷婷 -王 亨 王 彬 荣 茂
(1. 湖北省食品质量安全监督检验研究院,湖北 武汉 430075;2. 湖北省食品质量安全检测工程技术研究中心,湖北 武汉 430075)
喹喔啉类药物是一类包含有喹喔啉-1,4-二氮氧结构[1]通过化学合成的专用兽用药物,因其具有良好的抗菌和促进生长的效果,被广泛添加于禽、畜和水产养殖饲料中[2],其中喹乙醇是喹喔啉类药物的典型代表[3]。动物基体对于喹乙醇有蓄积作用,并且会引起动物产生畸形,对人类更是有致突变和致癌的作用[4]。喹乙醇原药的性质不稳定,在体内代谢迅速,能产生多种代谢产物,其中的绝大多数性质也不稳定,无法被定量检测。3-甲基-喹喔啉-2-羧酸(MQCA)作为喹乙醇的主要代谢产物之一,因其代谢过程比较长,并且残留量相对稳定,现在通常都是通过测定MQCA的含量来间接反映喹乙醇的残留情况[5]。中国自2019年5月1日起停止经营、使用喹乙醇兽药的原料药及各种制剂[6],而早在1998年,喹乙醇就已经被欧盟禁止作为饲料添加剂[7]。时至今日,MQCA用很多检测方法可以检测[8-12],其中液相色谱法和液相—质谱/质谱为主流的检测方式。动物源食品基质复杂,含有大量的脂肪、蛋白质等物质,液相色谱法一般需要经过相对复杂的前处理,且定性难度较大,容易产生假阳性;而液相—质谱/质谱检出限低,抗干扰能力和定性能力较强,可以在降低检出限的同时避免假阳性的产生。MQCA的前处理方式通常为酶解和水解(酸解),酶解耗时长,酸解提取效率不高,后续净化效果不彻底,检出限高,灵敏度不足。采用液相—质谱/质谱检测也需要不断地改进优化前处理方法,缩短样品测定时间,减少基质效应,提高灵敏度的同时保证准确度,李佩佩等[13]用免疫亲和柱对MQCA进行特异性吸附,效果较好,但免疫亲和柱价格成本较高,且容易堵塞[14]。痕量污染物分析检测的重点是简化操作,减少干扰,以期提高灵敏度;而难点是分离分析物和杂质以及合适的净化方法[15]。张小军等[16]采用盐酸水解提取MQCA,阴离子固相萃取柱净化,对鱼肉等水产食品的水解程度可以达到很好的效果,但对于畜禽肉水解效果并不是很理想;吴玉杰等[17]对水解液进行反复的液液萃取,操作繁复,过程损失大。
试验拟建立碱水解提取,固相萃取法净化和液相串联质谱联用的3-甲基-喹喔啉-2-羧酸的分析方法,为快速检测和监管动物源性食品中3-甲基-喹喔啉-2-羧酸残留提供技术保障和参考。
1 试验部分
1.1 材料及试剂
鸡肉、猪肉、鱼:武汉市售;
阴性基质:通过国家标准方法测定验证;
3-甲基-喹噁啉-2-羧酸标准品:纯度均≥99%,德国Dr.Ehrenstorfer公司;
喹噁啉-2-羧酸-D4(quinoxaline-2-carboxylic-D4,QCA-D4)标准品:纯度≥99%,德国Witega公司;
正己烷、盐酸、氢氧化钠:分析纯,上海国药集团化学试剂有限公司;
乙腈、甲酸、甲醇、乙酸乙酯:质谱级,德国默克公司;
Oasis MAX固相萃取柱:美国Waters公司;
试验用水:去离子水(电阻率≥18.2 MΩ·cm),Millipore-Q超纯水仪自制。
1.2 仪器与设备
串联四级杆质谱仪:Xevo TQD型,美国Water公司;
液相色谱仪:ACQUITY UPLC型,美国Water公司;
高速离心机:Allegra X-15R型,美国Beckman Coulter有限公司;
电子分析天平:XS204、ME2002E型,梅特勒—托利多仪器(上海)有限公司;
旋转蒸发器:HEI-VAP/LR20型,德国Heidolph GmbH公司;
超声波清洗器:Elmasonic P型,德国艾尔玛公司;
漩涡混合仪:EOFO基础型,美国Talboys公司;
水浴氮吹仪:N-EVAP24型,美国Organomation公司。
1.3 标准溶液的配制
在两个10 mL容量瓶中分别准确称量10 mg MQCA和QCA-D4标准品,加入甲醇溶解并稀释至刻度,配制成质量浓度为1 mg/mL的MQCA标准储备液和1 mg/mL QCA-D4同位素内标储备液。放置于-20 ℃的冰箱,有效期3个月。根据需要,用甲醇将MQCA和QCA-D4标准储备液稀释成浓度均为1 μg/mL中间标准溶液。临用前,将中间标准溶液用0.1%甲酸水溶液配制成0.5~50.0 ng/mL 的同位素内标曲线,每毫升该标准工作液含有同位素内标5 ng。
1.4 样品处理
称取5.00 g捣碎并已均匀混合的样品到50 mL的聚丙烯离心管中,然后加入50 μL QCA-D4同位素内标标准溶液,加入2 mol/mL NaOH溶液20 mL,涡旋均匀,60 ℃ 水浴条件下水解1 h[18]。待水解液冷却至室温后,用10 mol/L HCl溶液将水解液pH值调至1.0±0.2,4 000 r/min 离心5 min,上清液转移至50 mL离心管中,加入20 mL正己烷,充分混合,4 000 r/min离心5 min,去除正己烷层。向离心管中加入乙酸乙酯15 mL,涡旋混匀器上涡旋3 min,4 000 r/min、4 ℃条件下离心5 min,将上层乙酸乙酯转移至50 mL鸡心瓶中。提取液用15 mL 乙酸乙酯复提一次,合并乙酸乙酯于同一鸡心瓶中,50 ℃下旋转蒸发至净干,用5 mL 2%氨水溶液溶解残渣,待净化。用3 mL甲醇和3 mL水活化Oasis MAX固相萃取柱后,所有待净化的液体均经过MAX固相萃取柱,等待样品溶液完全流出后,用3 mL 0.05 mol/L氢氧化钠溶液,水和甲醇溶液依次淋洗, 将流出液全部弃去。 加压吹干2 min,并用3 mL 2%的甲酸甲醇溶液洗脱,洗脱液收集于15 mL离心管中[19]。洗脱液置于50 ℃水浴下氮气吹干,将残余物用1.0 mL的0.1%甲酸水复溶,0.22 μm 有机滤膜过滤,供液相串联质谱检测。内标法定量。
1.5 液相色谱条件
色谱柱型号:Waters ACQUITY UPLC BEH C18色谱柱(2.1 mm×50 mm,1.7 μm);色谱柱温度:35 ℃;进样量:5 μL;流速:0.30 mL/min;流动相A:0.1%甲酸水溶液,流动相B:乙腈;梯度洗脱过程:0.0~1.0 min,98% A;1.0~2.5 min,98%~50% A;2.5~2.6 min,50% A;2.6~3.0 min,50%~10% A;3.0~3.2 min,10%~98% A;3.2~5.0 min,98% A。
1.6 质谱参考条件
离子模式:正离子模式;离子源类型:电喷雾离子源(ESI源);离子源温度:120 ℃;扫描方式:多反应监测(MRM)模式;毛细管电压:2.00 kV;去溶剂气:高纯N2,流速:800 L/h,温度:450 ℃;锥孔气:高纯N2,流速50 L/h;碰撞气:高纯Ar。
2 结果与分析
2.1 质谱条件的确定
分别在ESI+和ESI-模式下,根据目标化合物的性质和结构,选择响应最高的离子加和峰作为母离子,通过对比ESI+和ESI-模式下的母离子响应,确定试验在ESI+下进行。对选定的母离子加碰撞能量击碎,同时进行离子扫描,分析可能的断裂规律以及根据二级碎片离子扫描质谱图,分别选取离子丰度相对较强的一对碎片离子作为定量离子,次强的一对碎片离子作为定性离子,最后在多反应监测模式(MRM)下分别对锥孔电压、碰撞能量进行优化。对于同位素内标物质而言,仅需一个丰度相对最强的一对碎片离子即可。表1中显示了最终确定多反应监测条件的质谱参数。
表1 3-甲基-喹噁啉-2-羧酸和内标物的离子对及锥孔电压、碰撞能量†
Table 1 Ion pair and cone voltage and collision energy of MQCA and internal standard
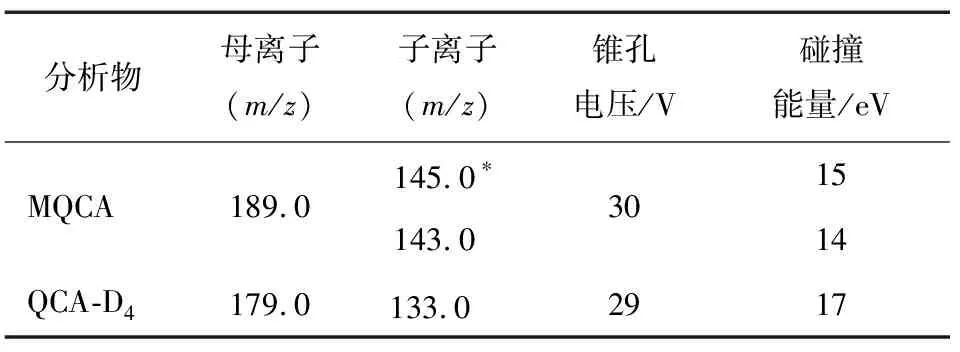
分析物母离子(m/z)子离子(m/z)锥孔电压/V碰撞能量/eVMQCA189.0145.0∗143.0301514QCA-D4179.0133.0∗2917
† 带*的离子为定量离子。
2.2 色谱条件的确定
MQCA为极性一般的弱酸性化合物,在C18柱上有好的保留效果[8],因此采用Waters ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm)的超高效液相色谱柱进行试验。同时,在正离子模式下以乙酸铵溶液作为流动相,会出现一定的离子抑制作用;而流动相中添加一定量的甲酸,减小pH值的同时,不仅可以改善峰型,还可以提高离子电离效率以及目标与杂质的分离度。通过对0.1%甲酸溶液与乙腈、甲醇分别作为有机相组成流动相的效果进行了比较。对于标准品而言,乙腈和甲醇均有较好的峰型,甲醇作为流动相时的峰宽要大于乙腈作为流动相时的峰宽;并且分离样品时,以乙腈作为流动相时不仅灵敏度略高,并且目标物和杂质的分离效果明显要比甲醇作为流动相时的目标物和杂质的分离效果好,因此试验选择流动相为乙腈—0.1%甲酸溶液。色谱条件经过优化后,MQCA的峰形得到改善,与基质中的杂质也有较好的分离度,峰型尖锐,整个分离过程可以在5 min内完成。标准溶液、空白基质样本及添加药物样本的选择离子流色谱图见图1~3。
2.3 水解方式的选择
水解方式一般为酶水解[20]、酸水解[21]、碱水解[22]3种,MQCA与其他兽残检测一样,也是先水解组织(破坏目标物与细胞的结合),再净化提取液,然后再检测净化液的流程。酶水解过程十分温和,需在(47±3) ℃振荡水浴中反应16 h以上,酶解过程耗时长且步骤繁琐;酸水解反应相对较温和,所以对于猪肉等样品,其水解效率不高。因为MQCA与组织主要是共价结合,采用酶解和酸解的方式并不能完全将MQCA从组织中解离出来[18]。强碱水解相对于酶解和酸解效果更好,反应通常较为剧烈,能在短时间内有效水解样品中的蛋白质并将MQCA从组织中解离出来。试验通过改变碱水解时间以及NaOH碱溶液的浓度,通过水解液状况评价水解效果。选择水解时间分别为20,40,60,80,100,120 min,NaOH溶液浓度分别采用0.2,0.5,1.0,2.0,5.0,10.0 mol/L,在60 ℃下对样品进行水解。通过对比发现,NaOH浓度太低,水解过程耗时长;NaOH浓度越高水解液相反会变得浓稠,不适合后续的提取净化过程。选用2 mol/L NaOH溶液水解60 min时,基质样品被完全水解,MQCA从样品中被完全释放出来,没有残渣剩余。因此,选择该条件进行后续试验。
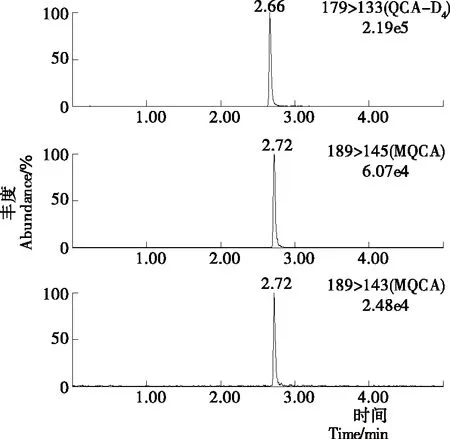
图1 标准溶液选择离子流色谱图
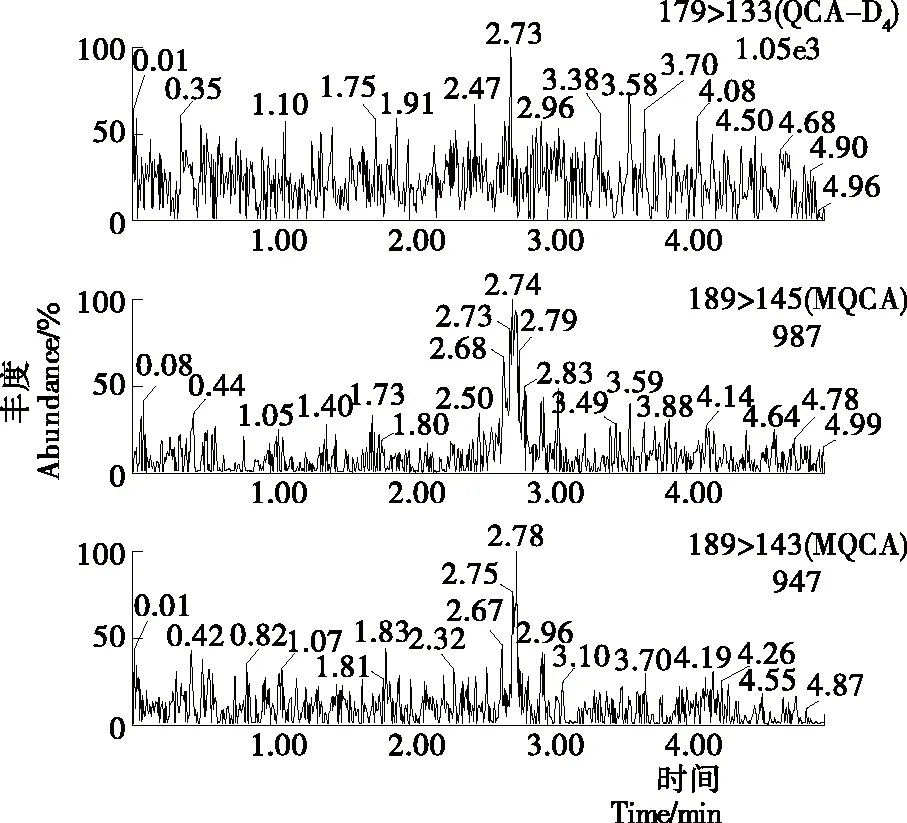
图2 空白基质样本选择离子流色谱图
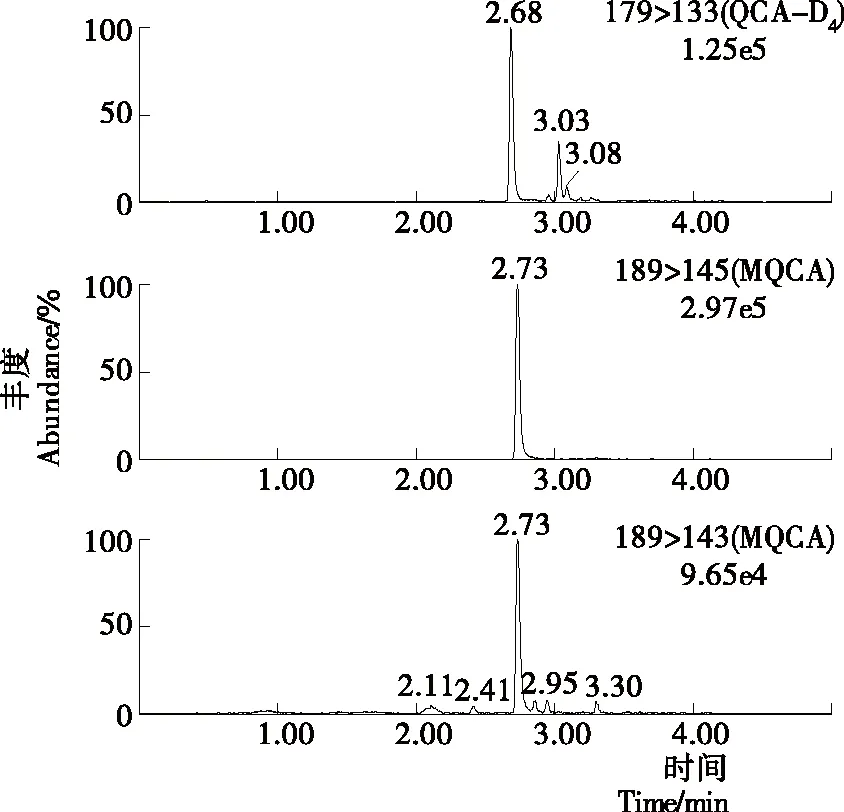
图3 添加药物样本的选择离子流色谱图
2.4 净化条件的确定
动物源样品通常包含大量的蛋白质、脂肪等内源性物质,直接检测必定会产生基质效应,对检测造成影响,对设备也会造成损害。常用净化的方式有固相萃取法和液液萃取法,试验采用液液萃取法与固相萃取法结合净化的方式。大多数的文献和国家标准都是选择的MAX固相萃取小柱[19,21-23],因为MQCA能解离形成负离子,可以与MAX上的季铵正离子发生相互作用结合。试验发现,水解液pH较大,滤液比较黏稠,若不经过处理直接过固相萃取柱容易堵塞固相萃取小柱筛板,使过柱流速变慢,导致后续净化过程无法进行,因此必须先通过液液萃取过程,将目标物萃取出来。由于MQCA呈弱酸性,水解液必须要经过酸化后MQCA才能形成分子形态,才能被乙酸乙酯萃取,并且MQCA分子在碱性条件下才能够离解形成负离子,与MAX上的季铵正离子才能充分键合。试验选择用浓盐酸对水解液进行酸化,强酸对蛋白起到一定的沉淀作用,乙酸乙酯萃取MQCA,浓缩乙酸乙酯后,用2%氨水溶液溶解残渣,通过MAX固相萃取小柱净化,整个过程符合阴离子固相萃取柱的阴离子交换作用的原理,可以达到理想的富集和净化的效果。
2.5 基质效应
将MQCA标准溶液分别用流动相和空白基质提取溶液稀释成0.5,1.0,2.0,5.0,10.0 ng/mL,均以峰面积为纵坐标,MQCA质量浓度为横坐标作图,即可分别得到标准工作曲线和基质曲线,通过两个曲线的斜率的比值可知,MQCA的基质曲线响应值约为标准曲线响应值的60%,说明MQCA在所验证基质中有一定程度的离子抑制效应,检测过程中的基质抑制效应和试验提取过程中的损耗会对定量带来误差。为了定量更加精准,在实际样品检测中既可采取基质匹配外标曲线校正法,也能采用同位素内标标准曲线法进行定量。鉴于基质匹配外表曲线校正法的操作相对比较麻烦,试验采用同位素内标标准曲线法作为最终的定量方法,MQCA和QCA化学性质相近,参考相关标准和文献[19,23-24],选择QCA-D4作为内标。
2.6 线性关系和检出限
根据上述所确定的液相条件、质谱参数及构建的MQCA残留的检测分析方法,对配制的0.5~50.0 ng/mL的MQCA系列标准工作曲线进行方法学指标分析。结果表明,在0.5~50.0 ng/mL浓度范围内,MQCA呈良好的线性关系,拟合曲线为y=0.659 6x+0.085 5(R2=0.999 7)。取阴性样品,通过加入低浓度MQCA标准溶液制得浓度含量低的加标样品,按照1.4方法对样品进行处理,以3倍信噪比计算方法的检出限,根据结果,确定此方法MQCA检出限为0.1 μg/kg (S/N=3)。
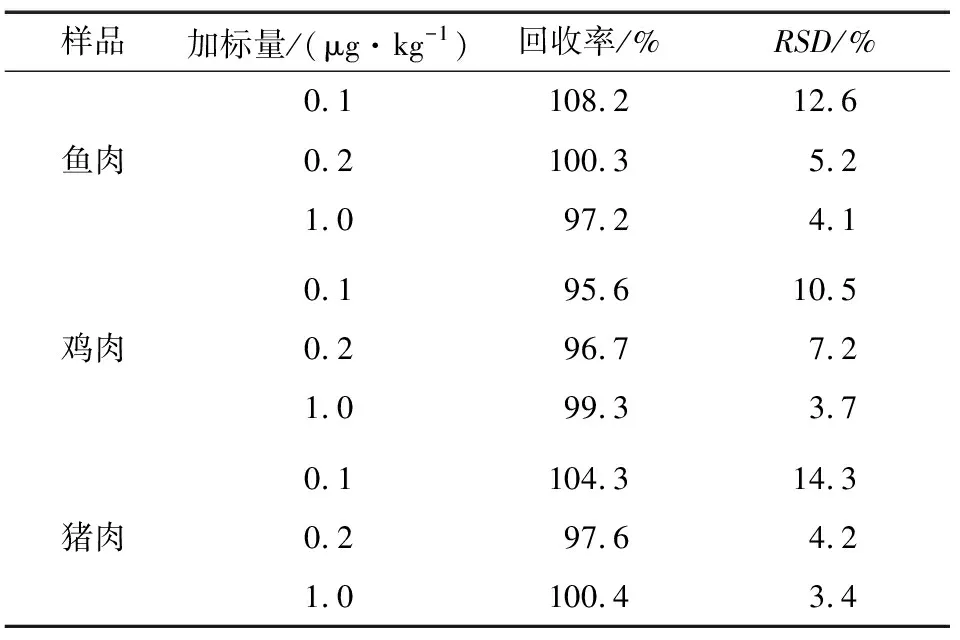
2.7 回收率和精密度
分别准确称取5.00 g猪肉、鸡肉、鱼肉捣碎样品(阴性),分别加入MQCA和QCA-D4标准溶液,得到0.1,0.2,1.0 μg/kg 3个浓度添加水平的加标样,每个浓度水平做6个平行。所得样品按照1.4方法处理,通过测定浓度与添加浓度之比计算回收率,并通过6次测定的回收率得出相对标准偏差。各基质加标回收率和相对标准偏差见表2。由表2可知,动物源食品中目标化合物的回收率为95.6%~108.2%,相对标准偏差为3.4%~14.3%(n=6)。
表2鱼,鸡肉和猪肉中MQCA的加标回收率和相对标准偏差
Table 2 Additive recovery and relative standard deviation of MQCA in fish,chicken and pork(n=6)
样品加标量/(μg·kg-1)回收率/%RSD/%0.1108.212.6鱼肉0.2100.35.21.097.24.10.195.610.5鸡肉0.296.77.21.099.33.70.1104.314.3猪肉0.297.64.21.0100.43.4
2.8 市售动物源食品的测定
采用试验方法对流通市场中的30批动物源食品样品(鱼、鸡肉、猪肉样品各10个)进行检测,结果表明,鱼肉和鸡肉样品中均未检出MQCA;有2个批次的猪肉中检出了MQCA,其残留量分别为0.23,3.08 μg/kg(根据GB/T 20746—2006国家标准方法测定的结果则为未检出和2.87 μg/kg)。综上可知,喹乙醇药物在中国仍在使用并存在组织残留的风险,在一定程度上对食品安全造成了威胁。渔业养殖和禽类养殖过程中对于喹喔啉类药物的使用和监管有了一定的成效;而由于喹乙醇代谢物在猪肉中存在允许限量,猪饲料里面允许加入适量的喹乙醇,所以猪肉中相对检出率要高。
3 结论
通过在碱性环境下水解提取样品组织,MQCA能够有效地从组织中游离,通过调节pH值在酸性介质下液液萃取提取MQCA,经过混合型强阴离子交换小柱净化等样品前处理技术除去大量杂质,减少基质效应,建立了一种快速、准确检测喹乙醇代谢物MQCA在动物源性食品中残留量的LC-MS/MS方法。该方法分离效果良好,操作简单快捷,准确度高,精密度较好。在0.1~1.0 μg/kg的添加水平范围内的平均回收率为95.6%~108.2%;相对标准偏差为3.4%~14.3%(n=6)。该方法适合动物源食品中MQCA残留的快速定量定性分析。运用高灵敏度和强特异性的免疫学方法的对大批量样品进行盲筛,以及结合液相串联质谱的强定性定量能力,发展基于新型材料更便捷的前处理方法,是今后亟需探索和发展的新方向。
