UPLC-PDA法同时测定连翘中4种成分的含量
2019-12-19张晓燕熊先明余世荣
刘 芳,魏 娟,,张晓燕,,熊先明,祝 宇,,余世荣*
(1.十堰市人民医院(湖北医药学院附属人民医院)药学部,湖北 十堰 442000;2.武当特色中药研究湖北省重点实验室,湖北 十堰 442000;3.兵兵宏康中药饮片(十堰)有限公司;湖北 十堰 442510)
连翘是木犀科植物连翘Forsythia suspense(Thunb.) Vahl的干燥果实,具有清热解毒、消肿散结、疏散风热的功效,按采收期的不同分为分青翘和老翘[1]。主要含苯乙醇苷类、黄酮类、木脂素类、萜类、挥发油类等化合物[2-5]。《中国药典》2015年版中规定以代表性的苯乙醇苷(连翘酯苷A)和木脂素类化合物(连翘苷)为连翘药材的质量评价指标。然而根据中药具有多成分、多靶点的特点,单一的成分很难全面反映连翘药材质量,采用多个指标性成分进行质量评价更具有科学性和全面性。
现代药理研究表明,槲皮素具有抗心肌缺血再灌注损伤、抗炎、抗肿瘤、抗高尿酸、抗高血糖等作用[6-10]。连翘酯苷B具有显著的抗炎抑菌、抗氧化的作用[11-12]。因此将其作为连翘的指标性成分进行研究,补充完善药典中连翘药材的质量评价指标。
1 仪器与试药
1.1 仪器
Waters Acquity H CLASS超高效液相色谱仪(美国Waters公司);CP214电子分析天平(奥豪斯仪器有限公司);H1650-W微量台式高速离心机(湖南湘仪实验仪器开发有限公司);2013QT超声波清洗器(北京碳氢超声波清洗机有限公司)。
1.2 试药
连翘苷,连翘酯苷A,连翘酯苷B,槲皮素对照品(四川省维克奇生物科技有限公司,批号wkp18012210,wkp19010907,wkp18100801,wkp19011509,纯度≥98 %);数批连翘(青翘)样品采自湖北省十堰市郧县十字沟(批号:20180801, 20180802,20180803),经湖北中医药大学陈科力教授鉴定为正品;乙腈,甲醇为色谱纯,水为双重蒸馏水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Acquity UPLC BEH C18柱(2.1 mm×100 mm,1.7 μm),预柱(1.7 μm,VANGUARD);以0.2 %的甲酸溶液为流动相A,乙腈为流动相B,梯度洗脱:0~15 min,90 %~70 %A;15~25 min ,70 %~30 % A;柱温:35 ℃;流速:0.3 ml/min;在330 nm波长下测定连翘酯苷B和连翘酯苷A的含量,在277 nm波长下测定连翘苷含量,在254 nm波长下测定槲皮素含量。
2.2 供试品溶液的制备
取样品细粉(过3号筛,50目)约1 g,精密称定,于50 ml具塞锥形瓶中,精密加入甲醇15 ml,称定重量,浸渍过夜,超声处理(250 W,40 kHz)25 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液5 ml置于干锅中蒸至近干,加入0.5 g中性氧化铝拌匀烘干,加在中性氧化铝柱(100~120目,1 g,内径为1~1.5 cm)上,用70 %乙醇80 ml洗脱,收集洗脱液,浓缩至干,残渣用50 %甲醇溶解,转移至5 ml量瓶,并稀释至刻度,摇匀,离心,取上清即得。
2.3 混合对照品溶液的制备
精密称取连翘酯苷B、连翘酯苷A、连翘苷、槲皮素对照品,加70 %的甲醇制成每1 ml分别含0.42,0.92,0.51,0.49 mg混合对照品溶液,摇匀,低温避光保存备用。
2.4 方法学考察
2.4.1 专属性试验 取连翘样品,按照2.2项方法制成供试品溶液,按2.1项色谱条件进样分析,得到样品的UPLC色谱图。参照4个对照品的色谱行为及其DAD紫外检测光谱图,结果见图1。在样品色谱图上对其峰进行指认,标定了4个成分,它们分别是图1中连翘酯苷B、连翘酯苷A、连翘苷和槲皮素。
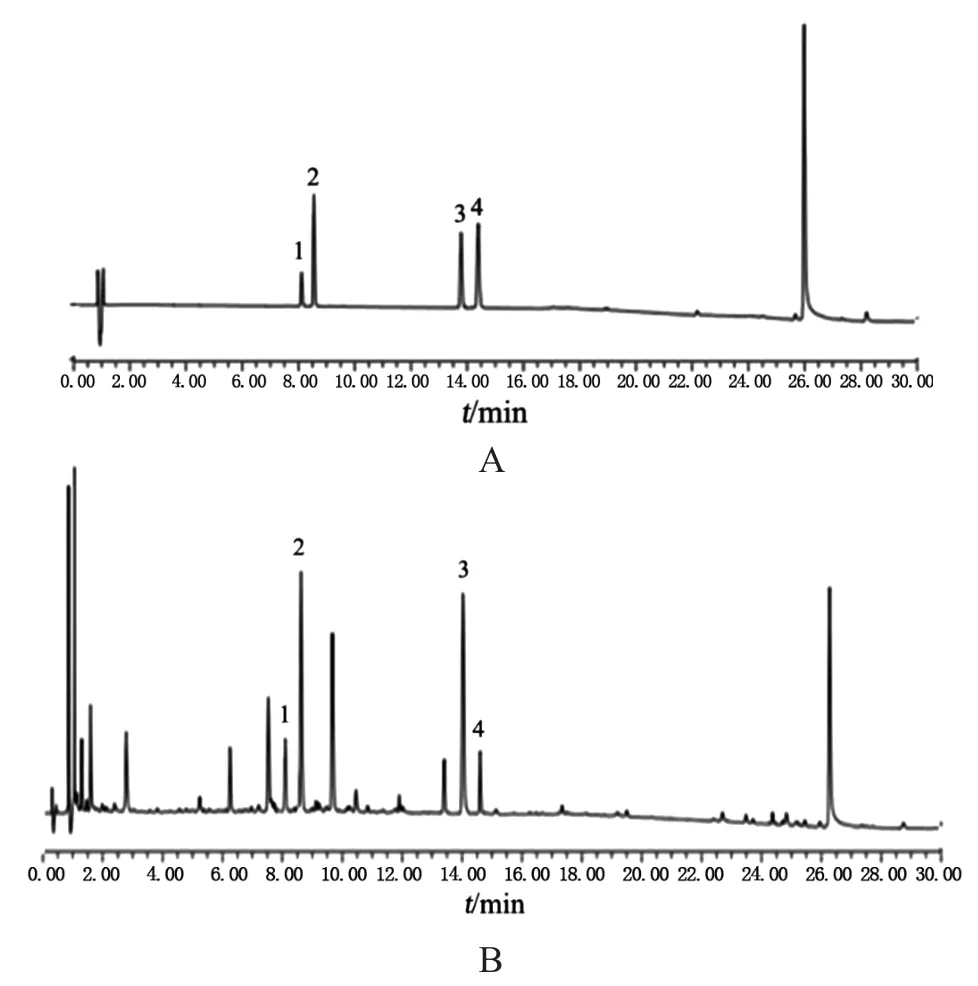
图1 UPLC色谱图
2.4.2 线性关系考察 为分析各药材标定成分的含量分布,按如下方法研究了各标定成分的线性回归分析参数:为分析各药材标定成分的含量分布,按如下方法研究了各标定成分的线性回归分析参数:精密吸取连翘酯苷B储备液(0.42 mg/ml)1,2,3,4,5 μl;连翘酯苷A储备液(0.92 mg/ml)1,2,3,4,5 μl;连翘苷储备液(0.51 mg/ml)2,3,4,5,6 μl;槲皮素储备液(0.49 mg/ml)0.2,1.4,2.6,3.8,5 μl,按2.1项下色谱条件进样测定,以进样量(μg)为横坐标,峰面积(A)为纵坐标进行线性回归,各成分回归方程分析参数见表1。结果表明连翘酯苷B、连翘酯苷A、连翘苷和槲皮素分别在进样量为0.42~2.10,0.92~4.60,1.02~3.06,0.098~2.45 μg范围内与峰面积线性关系良好。
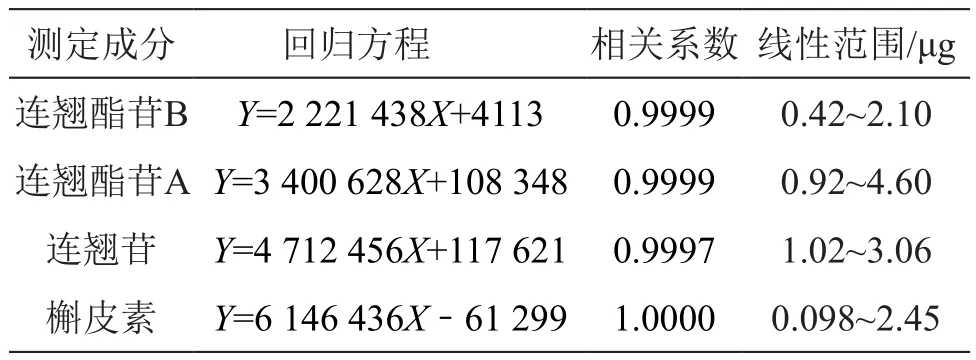
表1 4种成分线性关系考察试验数据
2.4.3 精密度试验 精密吸取混合对照品溶液,按照2.1项下色谱条件连续进样6次,分别记录其保留时间和峰面积,结果表明峰面积的RSD分别为0.28 %,0.73 %,1.35 %和0.57 %,表明仪器精密度良好。
2.4.4 稳定性试验 取同一供试品溶液(批号20180801),分别于室温下放置0,2,4,6,8,12,24 h,按照2.1项下色谱条件进样测定,记录其峰面积并计算RSD,结果表明其RSD分别为0.61 %,0.93 %,1.57 %和1.74 %,表明供试品溶液在24 h内稳定。
2.4.5 重复性试验 取同一批样品(批号20180801),按2.2项下方法平行制备供试品溶液6份,按2.1项下色谱条件分别进样测定,并计算4种成分含量,其RSD分别为1.81 %,2.04 %,2.61 %和2.39 %,表明该方法重复性良好。
2.4.6 加样回收率试验 取已知含量的连翘粉末样品(批号20180801)6份,各约1 g,精密称定,分别精密加入15ml连翘酯苷B(0.42 mg/ml)、连翘酯苷A(0.92 mg/ml)、连翘苷(0.51 mg/ml)和7.65 ml槲皮素(0.49 mg/ml),按2.2项下方法制备供试品溶液,再按2.1项下色谱条件进样测定,记录峰面积并计算回收率,结果见表1。

表1 连翘中4种成分的加样回收率试验结果
2.5 样品含量测定
精密称取3批连翘样品,按2.2项下方法制备供试品溶液,并按2.1项下色谱条件测定4种成分的含量,结果见表3。
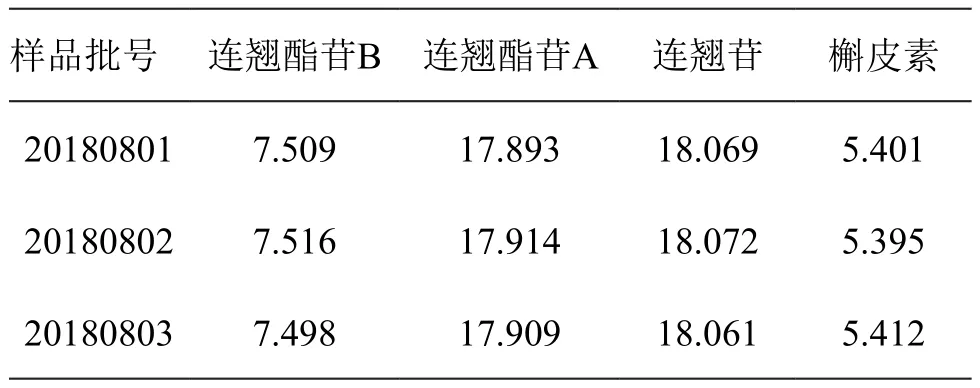
表3 4种成分含量测定结果/mg·g-1(n=3)
3 讨论
3.1 提取方法的选择
比较了超声提取、加热回流提取和过中性氧化铝柱子提取3种提取方式,发现用第3种提取方式得到的色谱图峰数较多,能同时检测到4种成分,峰形较好,分离状况令人满意,故选择过中性氧化铝柱子的提取方式。
3.2 波长的选择
用PDA检测器对样品在210~400 nm进行全波长扫描,发现连翘酯苷B和连翘酯苷A最大波长为330 nm,连翘苷和槲皮素分别在277 nm和254 nm波长下有最大吸收,分别在最大吸收波长下测定4种成分的含量。
3.3 流动相的选择
分别考察了乙腈-水、乙腈-冰醋酸、乙腈-磷酸、乙腈-甲酸为流动相时色谱峰的分离效果。结果,以乙腈-0.2 %甲酸溶液为流动相时分离效果最佳,色谱峰的分离度及峰形均良好,故最终选择乙腈-0.2 %甲酸溶液为流动相进行梯度洗脱。
3.4 UPLC与HPLC色谱的对比
对UPLC和HPLC的供试品色谱图进行比较发现,UPLC具有更高的分离度、灵敏度和更快的分离速度,单位时间内能提供更多的信息量,同时更节省流动相。
本文采用UPLC法同时测定连翘药材中4种成分含量,避免了单个成分测定带来的繁琐步骤,减少了流动相的配制和供试品的制备步骤,有效的提高了分析速度,缩短分析周期。该方法简便、快捷、准确且灵敏度高,可为连翘质量评价的科学性和全面性提供一定的参考。
