不同方式急性运动结合二甲双胍改善2型糖尿病小鼠血糖稳态及肝脏糖异生的作用
2019-12-17沈文清张强张怡漆正堂孙易丁树哲
沈文清 张强 张怡 漆正堂 孙易 丁树哲
摘 要:探讨不同方式急性运动结合二甲双胍对2型糖尿病小鼠血糖稳态及肝脏糖异生的作用,从运动中血糖变化、肝脏糖异生及调控因子相关mRNA表达水平的角度为治疗2型糖尿病提供新的运动处方和研究靶点。方法:采用4周高脂膳食结合链脲佐菌素(STZ,100 mg/kg)方法構建2型糖尿病小鼠模型,建模成功后随机分为NC、NCR、NCE、DC、DCR、DCE、HMC、HMR和HME共9组,每组8只小鼠。HMC、HMR和HME小鼠在末次运动前腹腔注射200 mg/kg HCloMetformin溶液,NC和DC组小鼠相应腹腔注射0.9%生理盐水。NCR、DCR和HMR小鼠进行急性抗阻运动;NCE、DCE和HME小鼠进行急性耐力运动。末次运动结束后3 h处死小鼠并取样。采用ELISA及RT-PCR技术检测相关血清指标和相关基因mRNA表达。结果:4周高脂膳食结合一次性腹腔注射STZ(100 mg/kg)成功构建2型糖尿病小鼠模型;相比DCE和DCR小鼠,HME和HMR小鼠血糖值和血糖波动幅度都显著下降;相比于HMC小鼠,HMR和HME小鼠附睾白色脂肪组织重量百分比、血清葡萄糖、血清TG和肝脏乳酸/丙酮酸浓度比值都显著下降,HMR组小鼠GSP和血清T-CHO显著降低,而HMR和HME小鼠肝糖原含量显著升高;相比HMC小鼠,HMR和HME小鼠肝脏糖异生相关因子PEPCK、G6pc和Gck mRNA表达显著升高,肝脏FBP和GLUT2 mRNA表达显著降低;相比于HMC小鼠,HMR和HME小鼠调控肝脏糖异生相关因子AMPKα2、CREB、PGC-1α、Keap1和Nrf2 mRNA表达都显著升高,且HMR小鼠肝脏AMPKα1 mRNA表达显著升高。结论:急性抗阻运动和急性耐力运动结合二甲双胍均可改善2型糖尿病小鼠运动中血糖的稳态,但急性抗阻运动结合二甲双胍能更有效改善2型糖尿病小鼠血糖稳态和肝脏糖异生情况。其中机制可能为不同方式的急性运动结合200 mg/kg剂量的二甲双胍可显著增加2型糖尿病小鼠肝脏AMPK-CREB-PGC-1α-PEPCK/G6Pase/GLUT2信号通路mRNA的表达。
关键词:急性抗阻运动;急性耐力运动;二甲双胍;血糖稳态;肝脏糖异生
中图分类号:G 804.5 学科代码:040302 文献标识码:A
Abstract:Objective: The purpose of the present study was to investigate the effects of different acute exercise combined with metformin on glucose homeostasis and hepatic gluconeogenesis in type 2 diabetic mice, hoping to provide new exercise treatment prescriptions and study protein targets for curing type 2 diabetes from the perspective of the glucose homeostasis in acute exercise, hepatic gluconeogenesis and regulatory factors relative mRNA level expression. Methods: The type 2 diabetic mice were established by feeding high fat diet and intraperitoneal injection with STZ(Streptozotocin, 100mg/kg). These mice were divided into 9 groups randomly, such as Normal Control group(NC), Normal Control Resistance Exercise group(NCR), Normal Control Endurance Exercise group(NCE), Diabetic Control group (DC), Diabetic Control Resistance Exercise (DCR), Diabetic Control Endurance Exercise(DCE), High dose of Metformin Control group(HMC), High dose of Metformin acute Resistance Exercise group(HMR) and High dose of Metformin acute Endurance Exercise group (HME), n=8 in each group. HMC, HMR and HME group mice were intraperitoneally injected with HCl·Metformin solution(200mg/kg weight) and NC and DC group mice were intraperitoneally injected with 0.9% saline. NCR, DCR and HMR group mice performed acute resistance exercise; NCE, DCE and HME group mice performed acute endurance exercise. All mice were sacrificed and selected with samples in 3 hours after acute exercise. ELISA was used for examining relative serum indicators and RT-PCR was used for testing relative gene mRNA expression. Results: The protocol of 4-week high fat diet and one time intraperitoneal injection with STZ successfully developed the model of type 2 diabetic mice. Compared to DCE and DCR group mice, blood glucose and the fluctuation change were significantly decreased in HME and HMR group mice. Compared to HMC group mice, the percentage of epididymal white adipose tissue content, serum glucose, serum TG and hepatic [lactate]/[pyruvate] ratio were all significantly decreased in HMR and HME group mice, glycosylated serum protein and serum T-CHO were both declined in HMR group mice, but the percentage of liver content was strongly increased in HMR and HME group mice. Compared to HMC group mice, relative hepatic gluconeogenic factors PEPCK, G6pc and Gck mRNA expression were clearly risen and hepatic FBP and GLUT2 mRNA expression were aggressively decreased. Compared to HMC group mice, relative hepatic gluconeogenic regulators AMPKα2, CREB, PGC-1α, Keap1 and Nrf2 mRNA expression were significantly increased in HMR and HME group mice and hepatic AMPKα1 mRNA expression was clearly risen in HMR group mice. Conclusions: Acute resistance exercise and acute endurance exercise combined with 200mg/kg dose of metformin can improve glucose homeostasis in type 2 diabetic mice, but acute resistance exercise combined with metformin is more effective than acute endurance exercise combined with metformin in improving glucose homeostasis and hepatic gluconeogenesis in type 2 diabetic mice, possibly via increasing relative mRNA expression of hepatic AMPK-CREB-PGC-1α-PEPCK/G6Pase/GLUT2 signaling pathways.
Keywords: acute resistance exercise; acute endurance exercise; metformin; glucose homeostasis; hepatic gluconeogenesis
二甲雙胍作为双胍类药物,提取自山羊豆碱,问世以来成为各国广泛使用的一线口服降糖类药物,主要治疗2型糖尿病及其并发症。近年有研究发现二甲双胍具有减肥、抗癌和延长寿命等功效。二甲双胍发挥作用的核心机制是改变细胞的能量代谢[1],有效降低2型糖尿病患者FPG(空腹血糖)、PPG(餐后血糖)和HbA1c(糖化血红蛋白)[2-4]。二甲双胍治疗2型糖尿病的作用机制包括:1)直接通过抑制肝脏糖异生降低肝脏葡萄糖和ATP生成[5];2)二甲双胍促进肌肉和脂肪组织对葡萄糖的摄取和利用,提高L6肌细胞中GLUT1/GLUT4表达比率,降低餐后血糖[6];3)减少小肠内葡萄糖吸收;4)通过抑制线粒体复合物I酶的活性和线粒体氧化磷酸化(OXPHOS),抑制ATP合成,激活AMPK活性,促进线粒体中脂肪酸β氧化,抑制脂肪合成和胰岛素抵抗作用[7];5)改善2型糖尿病患者胰岛素敏感性,提高胰岛β细胞对血糖的应答反应[8-9];6)GLP-1(胰高血糖素样肽-1)水平的提高[10]。
肝脏具有促进氧化代谢、合成肝糖原和分泌蛋白质等作用,对维持机体健康起到重要作用。二甲双胍对肝脏糖异生的作用与2型糖尿病的发病机制紧密相关,有研究发现,二甲双胍抑制线粒体复合物I酶活性,从而抑制胰高血糖素诱导的cAMP和蛋白激酶A(PKA)的信号传导[1],其他研究证实,二甲双胍抑制线粒体甘油磷酸脱氢酶[11],通过促进CREB结合蛋白(CBP)磷酸化抑制cAMP信号通路[12],不依赖LKB1/AMPK信号通路的PGC-1α mRNA过表达[5]和AMPK信号通路降低肝脏葡萄糖生成作用机制[13]。二甲双胍结合运动对2型糖尿病血糖稳态的影响,则既可能有促进作用[14-15],也可能有抑制作用[16-17]。关于二甲双胍结合运动对2型糖尿病小鼠肝脏糖异生作用的研究甚少。本研究试图通过探讨不同急性运动结合有效剂量二甲双胍(200 mg/kg)[18-19]对2型糖尿病小鼠血糖稳态及肝脏糖异生的作用,探索出有效改善2型糖尿病小鼠血糖稳态的运动结合二甲双胍处方及新的治疗靶点。
1 材料与方法
1.1 实验对象
清洁级4周龄C57BL/6J雄性小鼠84只,体质量为(12.39±0.13)g。其中:24只小鼠随机分为NC(普通对照组)、NCR(普通对照急性抗阻运动组)和NCE(普通对照急性耐力运动组),每组各8只,喂养普通饲料。60只小鼠进行2型糖尿病小鼠造模,采用4周高脂饲料膳食(45%脂肪含量),于小鼠9周龄一次性腹腔注射STZ(100 mg/kg),每隔3 d记录小鼠空腹血糖,注射后第9 d空腹血糖值高于11.1 mmol/L,即为造模成功。48只小鼠造模成功,随机分为6组:DC(糖尿病对照组)、DCR(糖尿病对照急性抗阻运动组)、DCE(糖尿病对照急性耐力运动组)、HMC(高剂量二甲双胍糖尿病对照组)、HMR(高剂量二甲双胍糖尿病急性抗阻运动组)和HME(高剂量二甲双胍糖尿病急性耐力运动组),每组各8只。实验中进行一次不同方式的急性运动干预。于运动前1 h,NCR、DCR、NCE和DCE小鼠分别腹腔注射0.9%生理盐水,HMR和HME小鼠腹腔注射200 mg/kg HCl·Metformin溶液,并且在不同时间段测量各组小鼠尾间静脉血糖值。NC、DC和HMC小鼠不做运动干预,NC和DC小鼠于末次急性运动干预前腹腔注射0.9%生理盐水,HMC、HMR和HME小鼠腹腔注射200 mg/kg HCl·Metformin溶液。
1.2 实验对象的运动方案
大量的研究探讨了长期运动干预结合二甲双胍对2型糖尿病血糖稳态和肝脏糖异生的作用,而急性运动干预结合二甲双胍对其血糖稳态及相关分子机制的研究较少。有研究发现,12周中等强度跑台运动训练结合二甲双胍治疗显著改善糖尿病大鼠的血糖稳态和抑制线粒体诱导的脂肪酸合成[20]。抗阻训练结合二甲双胍改善健康老年人肌肉中炎症反应,促进肌肉能量代谢[21]。因此,本研究采用急性抗阻和急性耐力运动结合二甲双胍的干预方式,观察2种运动模式能否改善2型糖尿病小鼠运动中高血糖现象和运动后抑制肝脏糖异生作用。急性抗阻运动,又名爬梯实验,每次自下而上于30 s内爬完坡度为85 °的1 m爬梯,5次/组,共3组;急性耐力运动,又名跑台实验,中等强度运动速度20 m/min,坡度为0 °,共50 min。
1.3 ELISA和RT-PCR
ELISA酶联免疫试剂盒测试小鼠血清相关指标,GSP、血清葡萄糖、血清胰岛素、血清TG和T-CHO。小鼠血糖采用尾间静脉血,以ACCU-CHEK Active罗氏活力型血糖仪进行检测。主要包括4个过程:1)提取肝脏组织mRNA。冰上称取肝脏组织约50 mg,使用Invitrogen Trizol方法提取肝脏mRNA。2)mRNA浓度和纯度检测。超微紫外/可见光分光光度计来测所提取的mRNA的浓度和纯度。3)mRNA反转录为cDNA。将提取的mRNA用TOYOBO FSQ101反转录试剂盒反转录为cDNA。4)RT-PCR扩增。ABI Step One型实时荧光定量PCR仪检测肝脏调控血糖稳态和肝脏糖异生相关基因,扩增所用的荧光染料为TOYOBO QPK201 SYBR Green,实验所用引物均由上海生物工程有限公司合成。
1.4 统计学分析
本研究各数据结果均采用M±SE的方式表示,统计软件为SPSS 22.0,组间分析采用单因素方差分析。作图使用Graph pad Prism5软件。P<0.05代表差异具有显著性,P<0.01代表差异具有非常显著性。
2 结果
2.1 高脂膳食结合STZ构建2型糖尿病小鼠模型
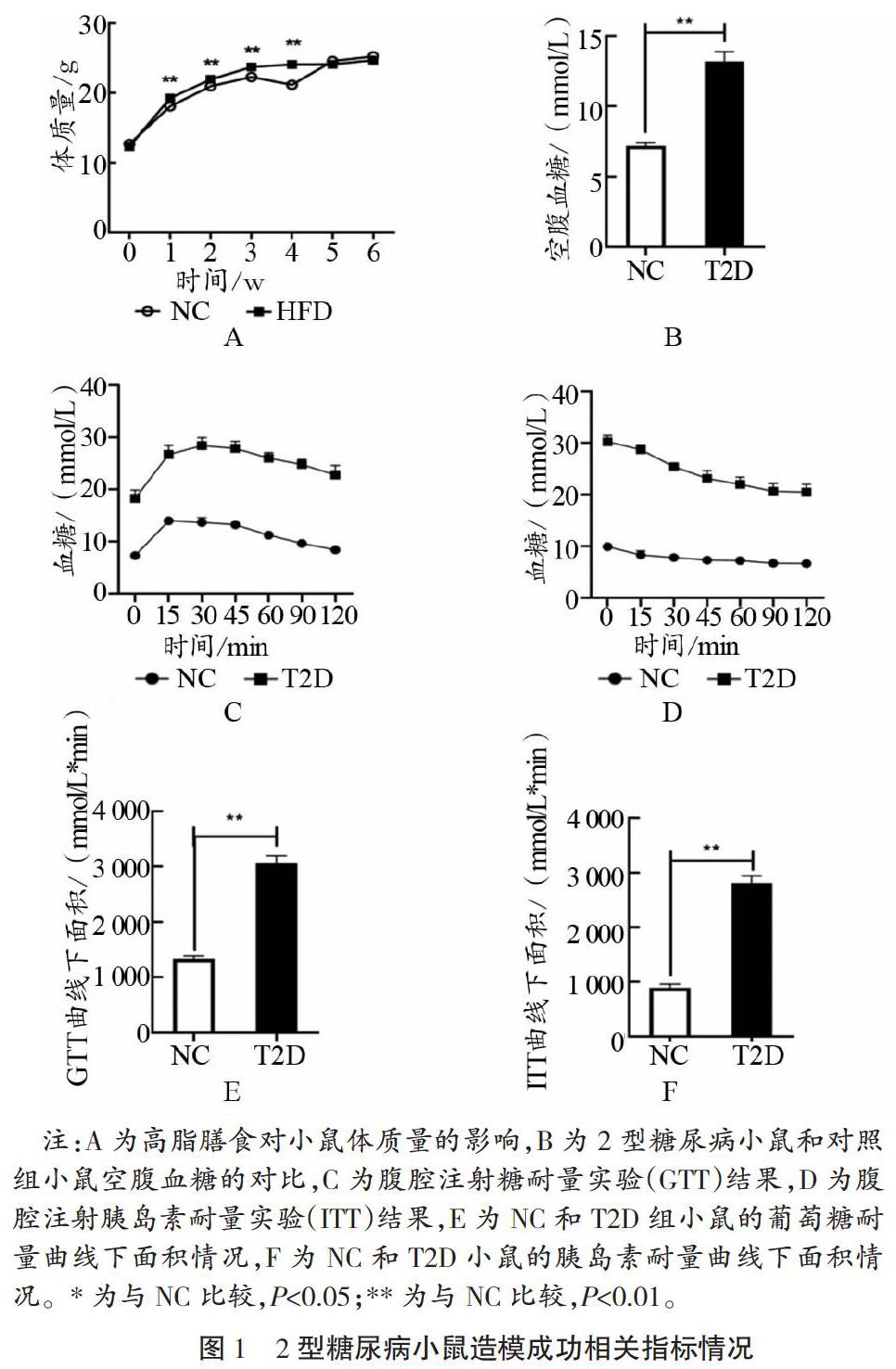
如图1所示, 4周高脂膳食(45%脂肪含量)使高脂膳食组(HFD)小鼠体质量在第1~4周均显著高于普通对照组(NC),P<0.01。HFD小鼠8周龄体质量显著比NC高13.8%。于小鼠9周龄空腹12 h后一次性腹腔注射STZ(链脲佐菌素,100 mg/kg),每隔3 d,监测小鼠的空腹血糖值,与STZ注射后第9 d,测得小鼠空腹血糖值>11.1 mmol/L,即2型糖尿病造模成功。2型糖尿病小鼠(T2D)空腹血糖值为(11.4±0.7)mmol/L,显著高于NC(P<0.01)。随后进行的葡萄糖耐量实验(IPGTT)和胰岛素耐量实验(ITT)都于小鼠空腹6 h后进行,测定小鼠尾间静脉血糖值,实验发现2型糖尿病小鼠血糖值在不同时间段都显著高于NC,且总体变化趋势一致。2型糖尿病小鼠葡萄糖AUC和胰岛素AUC(曲线下面积)都显著高于NC(P<0.01)。综上所述,4周高脂膳食结合腹腔注射STZ(100 mg/kg)成功构建2型糖尿病小鼠模型。
2.2 不同方式急性运动结合二甲双胍对2型糖尿病小鼠血糖稳态的影响
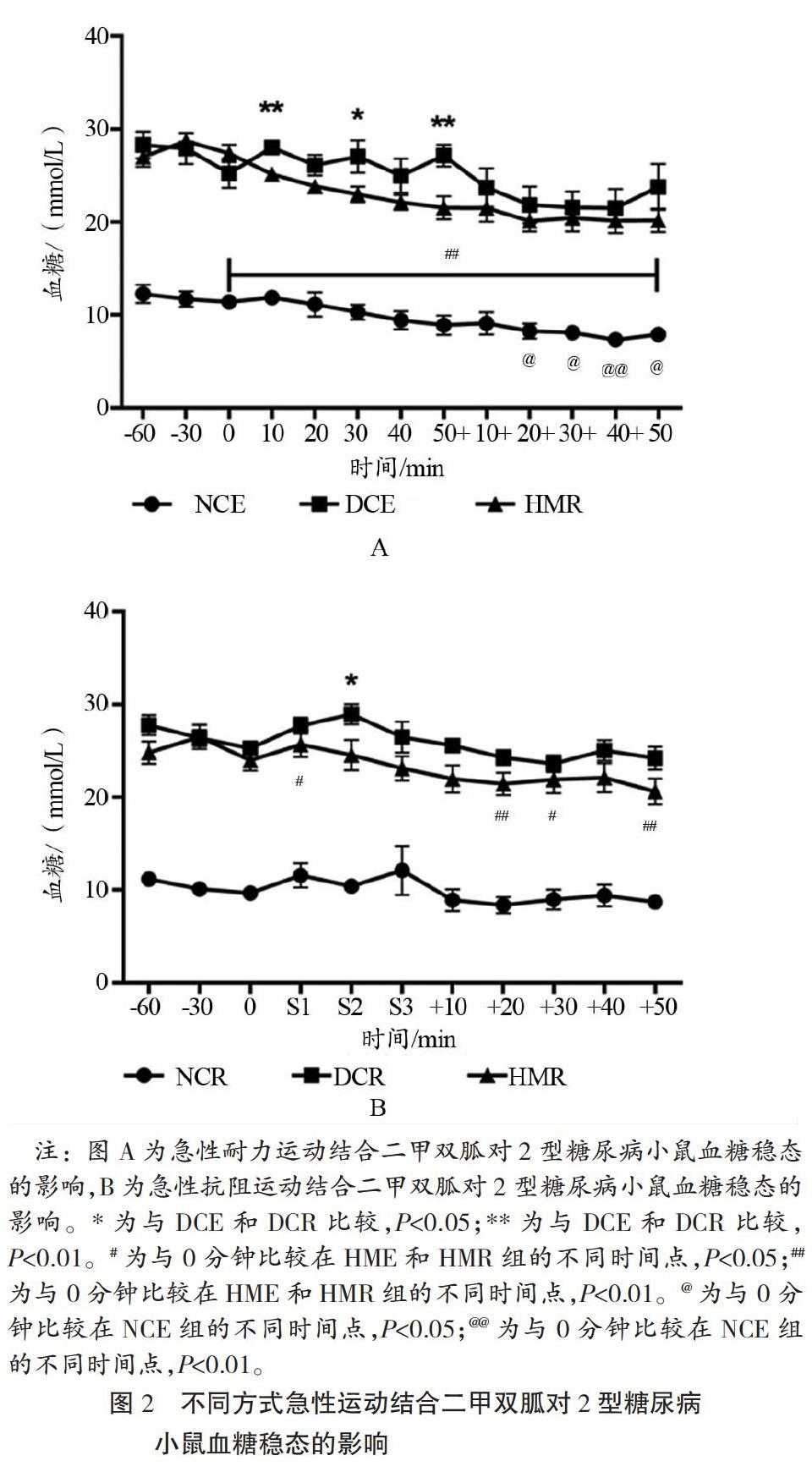
如图2所示,急性抗阻运动和急性耐力运动结合200 mg/kg剂量二甲双胍的干预模式均于运动干预前1 h针对不同组别小鼠分别腹腔注射生理盐水(NCE、DCE、NCR、DCR)和二甲双胍溶液(HME和HMR),在运动前、中、后不同时间点检测不同组别小鼠的尾间静脉血血糖值,急性耐力运动于运动前60 min每隔30 min测血糖,运动中50 min每隔10 min测血糖,运动后恢复阶段每隔10 min测血糖,急性抗阻运动血糖检测时间点于运动前和运动后恢复阶段与急性耐力运动一致,共进行3组抗阻运动(第1组S1、第2组S2和第3组S3),每组5次,每次自下而上于30 s内爬完坡度为85 °的1 m爬梯,每组抗阻运动结束后检测血糖值,如图2所示。相比于DCE和DCR小鼠,急性耐力运动和急性抗阻运动结合200 mg/kg剂量的二甲双胍都可降低2型糖尿病小鼠运动中血糖波动,促进其血糖稳态;因为运动中和运动后恢复阶段T2D小鼠血糖值均显著下降,并且血糖波动幅度降低。但是HME和HMR小鼠整体血糖水平都显著高于NCE和NCR(皆P<0.01)。
2.3 不同方式急性运动结合二甲双胍对2型糖尿病小鼠体成分的影响
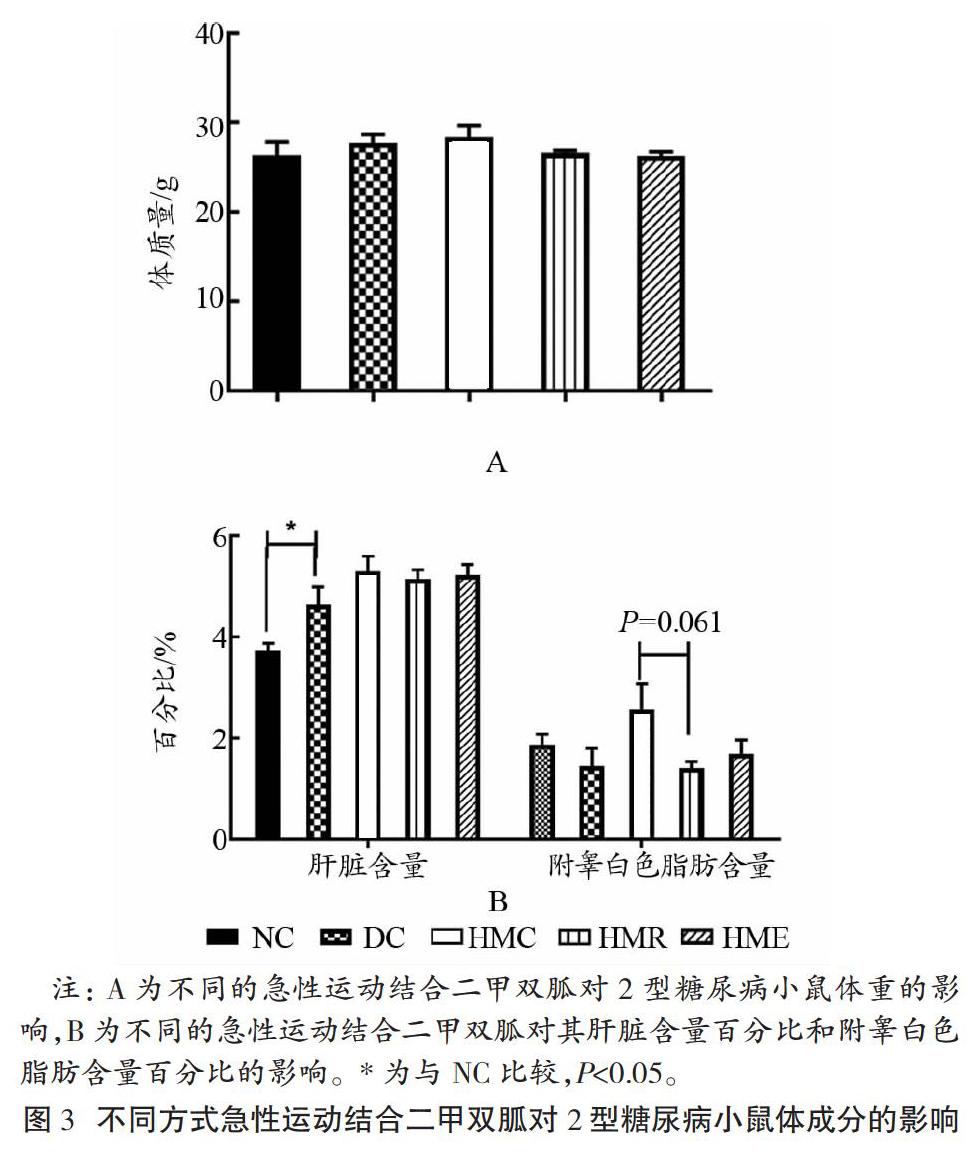
如图3所示,急性抗阻运动和急性耐力运动结合二甲双胍均未能显著改善2型糖尿病小鼠的体质量,但相较NC,DC小鼠肝脏含量百分比(肝指数)显著升高(P<0.05),其余各组未见显著差异。相较HMC,HMR组小鼠附睾白色脂肪含量百分比有下降的趋势(P=0.061)。
2.4 不同方式急性运动干预结合二甲双胍对2型糖尿病小鼠血液相关指标的影响
如图4所示,与NC比较,DC和HMC小鼠血清葡萄糖和糖化血清蛋白都显著升高,分别为P<0.01和P<0.05,同时DC和HMC小鼠血清TG(分别为P<0.01和P<0.05)和血清T-CHO都显著升高(分别为P<0.01和P<0.01)。与HMC比较,HMR小鼠血清葡萄糖和糖化血清蛋白都显著降低,分别为P<0.01和P<0.05,同时HMR小鼠血清TG和血清T-CHO都显著降低(P<0.01);与HMC比较,HME小鼠血清葡萄糖和血清TG都显著降低(分别为P<0.01和P<0.05),其他血清相关指标没有得到显著改善。与HMR比较,HME小鼠糖化血清蛋白显著升高(P<0.01),血清胰岛素显著降低(P<0.05)。
2.5 不同方式急性运动结合二甲双胍对2型糖尿病小鼠肝糖原合成和肝脏氧化还原状态的影响
如图5所示,与NC比较,DC和HMC小鼠肝糖原和丙酮酸含量显著升高(P<0.01),而DC小鼠肝脏乳酸/丙酮酸浓度比值显著降低(P<0.01)。与DC比较,HMC小鼠肝脏乳酸/丙酮酸浓度比值显著升高(P<0.01)。与HMC比较,HMR和HME小鼠肝糖原含量显著升高(P<0.01),而HMR小鼠肝脏乳酸含量、丙酮酸含量和肝脏乳酸/丙酮酸浓度比值都显著降低(P<0.01,P<0.05,P<0.05);而HME小鼠只有肝脏乳酸/丙酮酸浓度比值顯著降低(P<0.01)。相较HMR,HME小鼠肝脏丙酮酸含量显著升高(P<0.01)。
2.6 不同方式急性运动结合二甲双胍对2型糖尿病小鼠肝脏糖异生相关基因mRNA表达的影响
如图6所示,与DC比较,HMC小鼠肝脏PEPCK和G6pc mRNA表达显著下降(P<0.01),与葡萄糖转运相关的肝脏GLUT2 mRNA表达显著升高(P<0.05);但HMC小鼠肝脏Fbp (FBP1) mRNA表达未见显著性差异。与HMC比较,HMR和HME小鼠肝脏PEPCK和G6pc mRNA表达显著升高(P<0.01,P<0.05,P<0.01,P<0.01),肝脏Fbp mRNA表达显著下降(P<0.01),与葡萄糖转运相关的肝脏GLUT2 mRNA表达显著降低(P<0.01),肝脏Gck mRNA表达显著升高(P<0.01)。与HMR比较,HME小鼠肝脏G6pc mRNA表达显著升高(P<0.01)。
2.7 不同方式急性运动结合二甲双胍对2型糖尿病小鼠调控肝脏血糖稳态相关基因mRNA表达的影响
如图7所示,与NC比较,DC小鼠肝脏PGC-1α显著下降(P<0.01),其他与调控肝脏血糖稳态相关的基因mRNA表达未见显著差异。与DC比较,HMC小鼠肝脏AMPKα2、Nrf2、Keap1和CREB mRNA表达都显著下降(P<0.01,P<0.05,P<0.05,P<0.01),而肝脏AMPKα1和PGC-1α mRNA表达未见显著差异。与HMC比较,HMR和HME小鼠肝脏AMPKα2(P<0.05)、PGC-1α(P<0.05)、Nrf2(P<0.05)、Keap1(P<0.01和P<0.05)和CREB(P<0.01)mRNA表达都显著升高,只有HMR小鼠肝脏AMPKα1 mRNA表达显著升高(P<0.01),HME小鼠肝脏AMPKα1 mRNA表达未见显著差异。与HMR比较,HME小鼠肝脏PGC-1α显著升高(P<0.05)。
3 讨论
3.1 不同急性运动结合二甲双胍对2型糖尿病小鼠血糖稳态和体成分的影响
本研究发现,200mg/kg剂量的二甲双胍结合不同方式的急性运动干预方式(急性抗阻运动和急性耐力运动)都有助于降低2型糖尿病小鼠血糖值和血糖波动幅度。当前,关于急性运动干预结合二甲双胍对2型糖尿病血糖稳态作用的研究较少。6个月二甲双胍治疗可有效降低有高胰岛素血症和有2型糖尿病家族史的肥胖青少年的空腹血糖水平、血清胰岛素水平和BMI,显著提高其胰岛素敏感性,降低2型糖尿病的患病风险。二甲双胍还可改善2型糖尿病患者急性中等强度功率自行车运动中的血糖稳态[14]。长期运动干预结合二甲双胍服用和能量限制饮食结构有效降低肥胖青少年的2型糖尿病患病风险[22]。有研究发现,4周游泳运动结合450 mg/kg剂量的二甲双胍治疗显著降低胰岛素抵抗大鼠附睾白色脂肪重量百分比[23]。二甲双胍通过调控小鼠昼夜节律和AMPK-NAMPT-SIRT1信号通路抑制db/db小鼠白色脂肪组织堆积[24]。同时二甲双胍可促进人体脂肪组织脂联素的分泌和利用,促进脂代谢[25]。2型糖尿病的形成伴随脂代谢紊乱,运动结合二甲双胍可显著降低雌性糖尿病肥胖大鼠(ZDF)骨骼肌FAT/CD36丰富度、神经酰胺和二酰甘油(DAG)含量,促进游离脂肪酸氧化,降低脂肪堆积,从而抑制高脂膳食诱导的胰岛素抵抗和2型糖尿病的形成[26]。本研究认为,急性抗阻运动和急性耐力运动结合200 mg/kg剂量的二甲双胍虽未显著改善2型糖尿病小鼠的体质量,但DC小鼠肝脏质量百分比(肝脏质量/体质量,肝指数)显著高于NC。与HMC比较,HMR和HME小鼠附睾白色脂肪重量百分比显著降低,说明急性抗阻运动和急性耐力运动结合二甲双胍促进2型糖尿病小鼠脂肪酸氧化和脂代谢,降低其体脂百分比。
3.2 不同急性运动结合二甲双胍对2型糖尿病小鼠血清和肝脏调节血糖代谢相关指标的影响
4周游泳运动干预结合450 mg/kg剂量的二甲双胍可显著降低胰岛素抵抗大鼠的血清胰岛素水平[23]。短期中等强度游泳运动结合200 mg/kg剂量的二甲双胍可显著降低2型糖尿病妊娠期小鼠的血糖值和提高胰岛素敏感性,提高葡萄糖和脂肪氧化代谢水平[27]。急性和长期运动干预都间接促进2型糖尿病大鼠肝脏Leptin-AMPK-ACC信号通路的激活,增加胰岛素敏感性,然而只有长期运动干预可改善其血糖代谢和肝脏糖异生[28]。本研究认为,急性抗阻运动和急性耐力运动结合200 mg/kg剂量的二甲双胍均显著降低了2型糖尿病小鼠的血清葡萄糖、血清甘油三酯;与急性耐力运动结合二甲双胍相比,急性抗阻运动结合二甲双胍更能显著降低小鼠糖化血清蛋白和血清总胆固醇。运动中胰岛素水平的降低抑制胰高血糖素刺激的葡萄糖生成和糖异生,从而降低高血糖发生率和预防2型糖尿病[29]。但本研究未见到不同的运动干预方式结合200 mg/kg剂量的二甲双胍能降低2型糖尿病小鼠的血清胰岛素水平,可能因为运动干预时间和二甲双胍用药时间较短,不能诱导血清胰岛素水平下调。与HMR比较,HME小鼠的血清胰岛素显著降低,说明急性耐力运动结合二甲双胍优于急性抗阻运动结合二甲双胍对2型糖尿病小鼠血清胰岛素的调节作用。综上所述,不同方式的急性运动干预结合200 mg/kg剂量的二甲双胍均可改善2型糖尿病小鼠的血糖和血脂代谢,促进血糖稳态。
4周游泳运动结合二甲双胍、阿卡波糖治疗显著增加db/db小鼠的肝糖原含量[30]。研究发现,与HMC比较,急性抗阻运动和急性耐力运动结合200 mg/kg剂量的二甲双胍可显著提高2型糖尿病小鼠的肝糖原含量。同时急性游泳运动也显著增加糖尿病大鼠的肝糖原含量[31]。30 d自主转轮运动显著提高STZ诱导的糖尿病小鼠的肝糖原储备,并且抑制高血糖的发生[32]。2周中等强度有氧运动有效缓解糖尿病大鼠因急性运动引起的血糖升高,同时增加其肝糖原储备[33]。有研究表明,肝脏乳酸/丙酮酸浓度比值不仅反映肝脏氧化还原状态[34],也是肝脏糖异生的关键指标,反之浓度比值升高,会加快肝脏糖异生过程[35],肝脏乳酸/丙酮酸浓度比值的降低抑制胰高血糖素的功效,进而抑制高血糖的发生[36]。4周二甲双胍口服治疗和一次性静脉注射皆显著提高大鼠心脏和肝脏组织中乳酸/丙酮酸浓度比值,促进其氧化还原状态的改善[37]。单次低剂量二甲双胍(50 mg/kg)治疗使SD大鼠肝脏乳酸/丙酮酸浓度比值显著升高,改善肝脏的氧化还原状态[11]。二甲双胍除了对糖异生相关基因表达起抑制作用,另一重要原因是二甲双胍抑制肝脏乳酸的摄取和利用[38]。研究认为,与DC比较,单次200 mg/kg剂量的二甲双胍治疗显著提高2型糖尿病小鼠肝脏乳酸/丙酮酸浓度比值,改善肝脏细胞质氧化还原状态,一定程度上抑制2型糖尿病小鼠肝脏糖异生作用。与HMC比较,HMR小鼠肝脏乳酸、丙酮酸和肝脏乳酸/丙酮酸浓度比值显著降低,HME小鼠肝脏乳酸/丙酮酸浓度比值顯著降低;同时与HMR比较,HME小鼠肝脏丙酮酸含量显著升高,可能因为急性运动干预结合二甲双胍抑制肝细胞胞浆中的氧化还原状态,通过促进肝细胞线粒体中氧化还原状态抑制肝脏糖异生和促进血糖稳态。更多研究倾向于长期运动干预结合二甲双胍治疗2型糖尿病肝脏糖异生和血糖稳态,并且单一二甲双胍治疗在一定程度上削弱了运动促进机体氧化还原的作用[39]和运动抑制肥胖小鼠的肝脏糖异生作用[40]。与单一采用二甲双胍治疗相比,长期运动干预(体质量降低7%和每周150 min的体力活动)更有助于降低2型糖尿病的患病风险[41]。超重或肥胖人群通过一定强度的运动训练,可降低2型糖尿病的患病风险和体质量[42]。如何有机地结合运动干预和适宜剂量的二甲双胍,改善糖尿病小鼠或糖尿病患者运动中的血糖稳态,抑制其肝脏糖异生作用,有待进一步研究。
3.3 不同急性运动结合二甲双胍对2型糖尿病小鼠调节肝脏糖异生相关基因mRNA的影响
二甲双胍改善糖尿病状态下受干扰的膜流动性和蛋白质的构型,同时调节葡萄糖转运或代谢过程中所需正常功能蛋白质—蛋白质或蛋白质—脂肪的相互作用,细胞膜的变化可能对二甲双胍作用于胰岛素受体信号传导和各相关功能系统协同的作用[43]。运动能够降低高血糖,从源头抑制葡萄糖合成,并增加脂肪酸摄取和代谢,通过体内循环系统将多余的葡萄糖代谢。本研究中,与DC比较,HMC小鼠肝脏PEPCK和G6pc mRNA表达显著降低,GLUT2 mRNA表达显著升高,表明单一的二甲双胍治疗抑制2型糖尿病小鼠肝脏糖异生相关基因的转录水平,促进相关肝脏葡萄糖转运蛋白mRNA表达。与HMC比较,HMR和HME小鼠肝脏PEPCK、G6pc和Gck mRNA表达显著升高,肝脏Fbp和GLUT2 mRNA表达显著降低。在Ⅰ型糖尿病患者中发现,肝脏葡萄糖生成随运动强度的增加而增多,同时伴随糖异生作用的增强[44]。运动改善肝脏对胰岛素敏感性和对葡萄糖的摄取能力,长时间运动反向抑制肝脏葡萄糖输出[45],侧面反映急性运动干预可能促进肝脏葡萄糖输出,所以PEPCK和G6pc mRNA表达上调,而GLUT2 mRNA转录水平下调。本研究认为,急性运动干预结合200 mg/kg剂量的二甲双胍有效调节糖尿病小鼠运动中血糖稳态,但短期二甲双胍治疗结合急性运动干预未显著提高胰岛素抵抗患者的胰岛素敏感性,可能因为二者结合削弱运动对2型糖尿病的干预效果[20,46]。长期二甲双胍和/或运动干预可显著提高2型糖尿病前期患者的胰岛素敏感性,但存在二甲双胍钝化运动训练效益的现象[47]。AMPK是调节细胞能量稳态的重要调控因子,含有α、β、γ共3种亚基。二甲双胍和细胞内AMP/ATP浓度比值的增加激活AMPK活性,从而抑制肝脏糖异生过程和线粒体呼吸链复合物酶Ⅰ活性。AMPK激活剂二甲双胍和AICAR在一定程度上改善患有代谢综合征(包括2型糖尿病)小鼠的运动表现[48]。AMPK的活化促进脂代谢,调控2型糖尿病的胰岛素敏感性[1]。短期二甲双胍治疗未显著提高AMPKα2 KD(AMPKα2基因敲低)小鼠骨骼肌中胰岛素刺激的葡萄糖摄取,而长期二甲双胍治疗诱导依赖AMPK胰岛素刺激的骨骼肌葡萄糖摄取能力提高[49]。Bang等发现IPMK(肌醇多磷酸多激酶)在二甲双胍介导的LKB1/AMPK信号通路中发挥重要作用,可能是治疗2型糖尿病等代谢类疾病的新靶点[50]。
2型糖尿病的形成使多个组织器官的线粒体受损,包括线粒体形态学异常、线粒体含量减少、线粒体相关基因表达谱改变及酶活性降低[51-54]。2型糖尿病的形成与线粒体功能障碍息息相关,并且CREB、PGC-1α、Nrf2和Keap1是调控线粒体生物发生的重要基因。CREB是肝脏糖异生作用中PGC-1α的上游靶蛋白,空腹状态下CREB诱导metformin激活的PGC-1α表达,促进PGC-1α诱導肝脏糖异生作用的激活,调控血糖稳态[55]。二甲双胍诱导SHP基因表达的延迟效应,抑制依赖CREB的肝脏糖异生[56]。二甲双胍介导的CBPS436位点磷酸化抑制肝脏糖异生作用[57]。通过CBP磷酸化抑制cAMP信号通路,二甲双胍抑制肝脏糖异生[12]。胰岛素抑制肝脏葡萄糖生成,通过磷酸化CBP和使糖异生相关基因CREB-CBP复合物解体,其生物学作用通过二甲双胍促进p300结构性联接CREB[58]。有研究表明,PGC-1α是线粒体生物发生的有效诱导因子[59-60]。该作用通过PGC-1α与相关核转录因子(Nrf-1、Nrf-2和Tfam(线粒体转录因子A))结合和共激活,继而诱导与线粒体DNA复制相关的调控因子,发挥其调控作用[61]。相比野生型小鼠,PGC-1α基因过表达小鼠寿命更长,PGC-1α过表达促进线粒体功能的增强,同时增加胰岛素敏感性和降低氧化应激损伤[62]。本研究认为,与NC比较,DC小鼠肝脏PGC-1α mRNA表达显著降低。PGC-1α基因编码错义突变与2型糖尿病的形成显著相关,并且发现2型糖尿病患者体内PGC-1α基因表达和氧化磷酸化水平显著降低[51,63- 64]。急性抗阻运动和急性耐力运动结合二甲双胍显著增加2型糖尿病小鼠肝脏PGC-1α mRNA表达,促进线粒体呼吸链电子传递和氧化磷酸化,一定程度抑制肝脏糖异生。Keap1和Nrf2是抗氧化转录因子,降低2型糖尿病中胰岛β细胞和肝细胞ROS含量,促进其抗氧化、抗炎症功能和自噬作用[65]。Nrf2的活性由辅助因子Keap1氧化敏感的半胱氨酸残基控制,当Keap1被还原时,Nrf2发生蛋白酶体降解。半胱氨酸151位点氧化使Nrf2-Keap1复合物解体,使Keap1具有转录活性[66]。二甲双胍和运动都可以通过依赖Nrf2的方式激活抗氧化防御机制,促进2型糖尿病状态的改善[66]。本研究认为,与HMC比较,HMR和HME小鼠肝脏Nrf2和Keap1 mRNA表达显著升高。肠降血糖素相关的DPP4抑制剂激活Nrf2蛋白活性[66],抑制Nrf2蛋白活性削弱肿瘤转移[67],并减少细胞凋亡和自噬[68]。本研究认为,与DC比较,单次200 mg/kg剂量的二甲双胍治疗显著降低2型糖尿病小鼠肝脏CREB mRNA表达,抑制肝脏糖异生;与HMC比较,HMR和HME小鼠肝脏CREB mRNA表达显著升高。研究创新之处在于证实急性抗阻运动和急性耐力运动结合200 mg/kg剂量的二甲双胍皆可能通过显著增加2型糖尿病小鼠肝脏AMPK-CREB-PGC-1α-PEPCK/G6Pase/GLUT2信号通路mRNA表达改善其血糖稳态和调控肝脏糖异生作用,该通路可能是未来治疗2型糖尿病、调控血糖稳态和肝脏糖异生的有效靶点。
4 结论
急性抗阻运动和急性耐力运动结合200 mg/kg剂量的二甲双胍均可改善2型糖尿病小鼠血糖稳态,但急性抗阻运动结合二甲双胍可更有效改善2型糖尿病小鼠血糖稳态和肝脏糖异生,可能通过提高肝脏AMPK-CREB-PGC-1α-PEPCK/G6Pase/GLUT2信号通路mRNA的表达发挥作用。
參考文献
[1] PERNICOVA I, KORBONITS M. Metformin--mode of action and clinical implications for diabetes and cancer[J]. Nature reviews(Endocrinology), 2014, 10(3): 143.
[2] 中华医学会糖尿病学分会.中国2型糖尿病防治指南:2013年版[J].中国糖尿病杂志,2014,22(8):2.
[3] GARBER A J, DUNCAN T G, GOODMAN A M, et al. Efficacy of metformin in type II diabetes: results of a double-blind, placebo-controlled, dose-response trial[J]. The American Journal of Medicine, 1997, 103(6): 491.
[4] HOFFMANN J, SPENGLER M. Efficacy of 24-week monotherapy with acarbose, metformin, or placebo in dietary-treated NIDDM patients: the essen-II study[J]. The American Journal of Medicine, 1997, 103(6): 483.
[5] FORETZ M, HEBRARD S, LECLERC J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state[J]. The Journal of Clinical Investigation, 2010, 120(7): 2355.
[6] SARABIA V, LAM L, BURDETT E, et al. Glucose transport in human skeletal muscle cells in culture. Stimulation by insulin and metformin[J]. The Journal of Clinical Investigation, 1992, 90(4): 1386.
[7] ZHOU G, MYERS R, LI Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action[J]. The Journal of Clinical Investigation, 2001, 108(8): 1167.
[8] LUPI R, DEL G S, TELLINI C, et al. The biguanide compound metformin prevents desensitization of human pancreatic islets induced by high glucose[J]. European Journal of Pharmacology, 1999, 364(2-3): 205.
[9] MASINI M, ANELLO M, BUGLIANI M, et al. Prevention by metformin of alterations induced by chronic exposure to high glucose in human islet beta cells is associated with preserved ATP/ADP ratio[J]. Diabetes Research and Clinical Practice, 2014, 104(1): 163.
[10] CHO Y M, KIEFFER T J. New aspects of an old drug: metformin as a glucagon-like peptide 1 (GLP-1) enhancer and sensitiser[J]. Diabetologia, 2011, 54(2): 219.
[11] MADIRAJU A K, ERION D M, RAHIMI Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase[J]. Nature, 2014, 510(7506): 542.
[12] HE L, SABET A, DJEDJOS S, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein[J]. Cell, 2009, 137(4): 635.
[13] DUCA F A, COTE C D, RASMUSSEN B A, et al. Metformin activates a duodenal ampk-dependent pathway to lower hepatic glucose production in rats[J]. Nature Medicine, 2015, 21(5): 506.
[14] HANSEN M, PALSOE M K, HELGE J W, et al. The effect of metformin on glucose homeostasis during moderate exercise[J]. Diabetes Care, 2015, 38(2): 293.
[15] MALIN S K, BRAUN B. Impact of metformin on exercise-induced metabolic adaptations to lower type 2 diabetes risk[J]. Exercise and Sport Sciences Reviews, 2016, 44(1): 4.
[16] BOULE N G, ROBERT C, BELL G J, et al. Metformin and exercise in type 2 diabetes: examining treatment modality interactions[J]. Diabetes Care, 2011, 34(7): 1469.
[17] MYETTE-COTE E, TERADA T, BOULE N G. The effect of exercise with or without metformin on glucose profiles in type 2 diabetes: a pilot study[J]. Canadian Journal of Diabetes, 2016, 40(2): 173.
[18] 陳致瑜,潘润桑,罗振华,等.二甲双胍改善自发性2型糖尿病KKAy小鼠胰岛β细胞功能机制初探[J].贵州医科大学学报,2017,42(9):1016.
[19] 徐丽,李冬梅,孙慧萍,等.黄连素合用二甲双胍对糖尿病小鼠血糖的影响[J].实用药物与临床,2014,17(7):822.
[20] LINDEN M A, FLETCHER J A, MORRIS E M, et al. Combining metformin and aerobic exercise training in the treatment of type 2 diabetes and NAFLD in OLETF rats[J]. American Journal of Physiology(Endocrinology and metabolism), 2014, 306(3): 300.
[21] LONG D E, PECK B D, MARTZ J L, et al. Metformin to augment strength training effective response in seniors (masters): study protocol for a randomized controlled trial[J]. Trials, 2017, 18(1): 192.
[22] GARNETT S P, GOW M, HO M, et al. Improved insulin sensitivity and body composition, irrespective of macronutrient intake, after a 12 month intervention in adolescents with pre-diabetes; resist a randomised control trial[J]. BMC Pediatrics, 2014(14): 289.
[23] CHIEN K Y, HUANG C C, HSU K F, et al. Swim training reduces metformin levels in fructose-induced insulin resistant rats[J]. Journal of Pharmacy and Pharmaceutical Sciences, 2012, 15(1): 85.
[24] CATON P W, KIESWICH J, YAQOOB M M, et al. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice[J]. Diabetes, Obesity & Metabolism, 2011, 13(12): 1097.
[25] ZULIAN A, CANCELLO R, GIROLA A, et al. In vitro and in vivo effects of metformin on human adipose tissue adiponectin[J]. Obesity Facts, 2011, 4(1): 27.
[26] SMITH A C, MULLEN K L, JUNKIN K A, et al. Metformin and exercise reduce muscle FAT/CD36 and lipid accumulation and blunt the progression of high-fat diet-induced hyperglycemia[J]. American Journal of Physiology(Endocrinology and Metabolism), 2007, 293(1): 172.
[27] HUANG L, YUE P, WU X, et al. Combined intervention of swimming plus metformin ameliorates the insulin resistance and impaired lipid metabolism in murine gestational diabetes mellitus[J]. Plos One, 2018, 13(4): 195609.
[28] YI X, CAO S, CHANG B, et al. Effects of acute exercise and chronic exercise on the liver leptin-AMPK-ACC signaling pathway in rats with type 2 diabetes[J]. Journal of Diabetes Research, 2013(12): 946432.
[29] LAVOIE C, DUCROS F, BOURQUE J, et al. Glucose metabolism during exercise in man: the role of insulin and glucagon in the regulation of hepatic glucose production and gluconeogenesis[J]. Canadian Journal of Physiology Pharmacology, 1997, 75(1): 26.
[30] TANG T, REED M J. Exercise adds to metformin and acarbose efficacy in db/db mice[J]. Metabolism, 2001, 50(9): 1049.
[31] BICER M, AKIL M, AVUNDUK M C, et al. Interactive effects of melatonin, exercise and diabetes on liver glycogen levels[J]. Endokrynologia Polska, 2011, 62(3): 252.
[32] DE CARVALHO A K, DA S S, SERAFINI E, et al. Prior exercise training prevent hyperglycemia in STZ mice by increasing hepatic glycogen and mitochondrial function on skeletal muscle[J]. Journal of Cellular Biochemistry, 2017, 118(4): 678.
[33] MOTIANI K K, SAVOLAINEN A M, ESKELINEN J J, et al. Two weeks of moderate-intensity continuous training, but not high-intensity interval training, increases insulin-stimulated intestinal glucose uptake[J]. Journal of Applied Physiology (1985), 2017, 122(5): 1188.
[34] KREBS H A, GASCOYNE T. The redox state of the nicotinamide-adenine dinucleotides in rat liver homogenates[J]. The Biochemical Journal, 1968, 108(4): 513.
[35] SISTARE F D, HAYNES R J. The interaction between the cytosolic pyridine nucleotide redox potential and gluconeogenesis from lactate/pyruvate in isolated rat hepatocytes. Implications for investigations of hormone action[J]. The Journal of Biological Chemistry, 1985, 260(23): 12748.
[36] SUGANO T, SHIOTA M, TANAKA T, et al. Intracellular redox state and stimulation of gluconeogenesis by glucagon and norepinephrine in the perfused rat liver[J]. Journal of Biochemistry, 1980, 87(1): 153.
[37] LEWIS A J, MILLER J J, MCCALLUM C, et al. Assessment of metformin-induced changes in cardiac and hepatic redox state using hyperpolarized [1-13C] pyruvate[J]. Diabetes, 2016, 65(12): 3544.
[38] RADZIUK J, ZHANG Z, WIERNSPERGER N, et al. Effects of metformin on lactate uptake and gluconeogenesis in the perfused rat liver[J]. Diabetes, 1997, 46(9): 1406.
[39] WATSON J D. Type 2 diabetes as a redox disease[J]. Lancet, 2014, 383(9919): 841.
[40] SOUZA P L, ROPELLE E C, DE SOUZA C T, et al. Exercise training decreases mitogen-activated protein kinase phosphatase-3 expression and suppresses hepatic gluconeogenesis in obese mice[J]. The Journal of Physiology, 2014, 592(6): 1325.
[41] KNOWLER W C, BARRETT-CONNOR E, FOWLER S E, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin[J]. The New England Journal of Medicine, 2002, 346(6): 393.
[42] FLOREZ H, PAN Q, ACKERMANN R T, et al. Impact of lifestyle intervention and metformin on health-related quality of life: the diabetes prevention program randomized trial[J]. Journal of General Internal Medicine, 2012, 27(12): 1594.
[43] WIERNSPERGER N F. Membrane physiology as a basis for the cellular effects of metformin in insulin resistance and diabetes[J]. Diabetes & Metabolism, 1999, 25(2): 110.
[44] PETERSEN K F, PRICE T B, BERGERON R. Regulation of net hepatic glycogenolysis and gluconeogenesis during exercise: impact of type 1 diabetes[J]. The Journal of Clinical Endocrinology and Metabolism, 2004, 89(9): 4656.
[45] KOYAMA Y, GALASSETTI P, COKER R H, et al. Prior exercise and the response to insulin-induced hypoglycemia in the dog[J]. American Journal of Physiology(Endocrinology and Metabolism), 2002, 282(5): 1128.
[46] SHAROFF C G, HAGOBIAN T A, MALIN S K, et al. Combining short-term metformin treatment and one bout of exercise does not increase insulin action in insulin-resistant individuals[J]. American Journal of Physiology(Endocrinology and Metabolism), 2010, 298(4): 815.
[47] MALIN S K, GERBER R, CHIPKIN S R, et al. Independent and combined effects of exercise training and metformin on insulin sensitivity in individuals with prediabetes[J]. Diabetes Care, 2012, 35(1): 131.
[48] NARKAR V A, DOWNES M, YU R T, et al. AMPK and PPAR delta agonists are exercise mimetics[J]. Cell, 2008, 134(3): 405.
[49] KRISTENSEN J M, TREEBAK J T, SCHJERLING P, et al. Two weeks of metformin treatment induces AMPK-dependent enhancement of insulin-stimulated glucose uptake in mouse soleus muscle[J]. American Journal of Physiology(Endocrinology and Metabolism), 2014, 306(10): 1099.
[50] BANG S, CHEN Y, AHIMA R S, et al. Convergence of IPMK and LKB1-AMPK signaling pathways on metformin action[J]. Molecular Endocrinology, 2014, 28(7): 1186.
[51] MOOTHA V K, LINDGREN C M, ERIKSSON K F, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes[J]. Nature Genetics, 2003, 34(3): 267.
[52] MORINO K, PETERSEN K F, DUFOUR S, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents[J]. The Journal of Clinical Investigation, 2005, 115(12): 3587.
[53] PATTI M E, BUTTE A J, CRUNKHORN S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1[J]. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(14): 8466.
[54] RITOV V B, MENSHIKOVA E V, HE J, et al. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes[J]. Diabetes, 2005, 54(1): 8.
[55] HERZIG S, LONG F, JHALA U S, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1[J]. Nature, 2001, 413(6852): 179.
[56] LEE J M, SEO W Y, SONG K H, et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner[J]. The Journal of Biological Chemistry, 2010, 285(42): 32182.
[57] HE L, MENG S, GERMAIN-LEE E L, et al. Potential biomarker of metformin action[J]. The Journal of Endocrinology, 2014, 221(3): 363.
[58] HE L, NAIK K, MENG S, et al. Transcriptional co-activator p300 maintains basal hepatic gluconeogenesis[J]. The Journal of Biological Chemistry, 2012, 287(38): 32069.
[59] DILLON L M, REBELO A P, MORAES C T. The role of PGC-1 coactivators in aging skeletal muscle and heart[J]. IUBMB Life, 2012, 64(3): 231.
[60] SCARPULLA R C, VEGA R B, KELLY D P. Transcriptional integration of mitochondrial biogenesis[J]. Trends in Endocrinology and Metabolism: TEM, 2012, 23(9): 459.
[61] LARSSON N G, WANG J, WILHELMSSON H, et al. Mitochondrial transcription factor a is necessary for mtDNA maintenance and embryogenesis in mice[J]. Nature Genetics, 1998, 18(3): 231.
[62] WENZ T, ROSSI S G, ROTUNDO R L, et al. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging[J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(48): 20405.
[63] MOOTHA V K, LINDGREN C M, ERIKSSON K F, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes[J]. Nature Genetics, 2003, 34(3): 267.
[64] RUSSELL A P. PGC-1alpha and exercise: important partners in combating insulin resistance[J]. Current Diabetes Reviews, 2005, 1(2): 175.
[65] URUNO A, YAGISHITA Y, YAMAMOTO M. The keap1-Nrf2 system and diabetes mellitus[J]. Archives of Biochemistry and Biophysics, 2015(566): 76.
[66] TSCHOP M H, STUMVOLL M, RISTOW M. Opposing effects of antidiabetic interventions on malignant growth and metastasis[J]. Cell Metabolism, 2016, 23(6): 959.
[67] WANG H, LIU X, LONG M, et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis[J]. Science Translational Medicine, 2016, 8(334): 334.
[68] RAO V A, KLEIN S R, BONAR S J, et al. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone[J]. The Journal of Biological Chemistry, 2010, 285(45): 34447.
