热带植物基因组研究进展
2019-12-14李丹陈晓慧赖钟雄
李丹 陈晓慧 赖钟雄
摘 要 基于全基因组测序结果,探讨了包括热带水果香蕉(Musa spp.)、龙眼(Dimocarpus longan)、番木瓜(Carica papaya)、菠萝(Ananas comosus)、椰子(Cocos nucifera)、榴莲(Durio zibethinus),经济作物橡胶(Hevea brasiliensis)、木薯(Manihot esculenta)、枣椰(Phoenix dactylifera)、可可(Theobroma cacao)、油棕(Elaeis guineensis)、咖啡(Coffea canephora)以及药用植物铁皮石斛(Dendrobium officinale)在内的13种热带植物的全基因组测序的发展历程,并对热带植物基因组研究进行了概述。
关键词 热带植物;全基因组;第二代测序;遗传育种;功能基因中图分类号 Q943.2 文献标识码 A
Research Progresses of Tropical Plant Genome
LI Dan, CHEN Xiaohui*, LAI Zhongxiong**
Institute of Horticultural Biotechnology, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China
Abstract Based on genome-wide sequencing results, the development of genome-wide sequencing of 13 tropical plants including banana (Musaspp.), longan (Dimocarpus longan), papaya (Carica papaya), pineapple (Ananas comosus), coconut (Cocos nucifera), durian (Durio zibethinus), rubber (Hevea brasiliensis), cassava (Manihot esculenta), date palm (Phoenix dactylifera), cocoa (Theobroma cacao), oil palm(Elaeis guineensis), coffee (Coffea canephra) Tiepi-shihu (Dendrobium officinale) was discussed, and the tropical plant genome research was summarized.
Keywords tropical plants; whole genome; next generation sequencing; genetic breeding; functional genes
DOI10.3969/j.issn.1000-2561.2019.10.001
20世纪末,以Sanger技术为核心的第一代测序技术诞生,单链DNA噬菌体φX174全基因组序列的测定标志着人类正式步入基因组学时代[1]。第一代测序技术准确性高、序列读长可达1 kb,但其测序技术复杂、成本高、通量低,无法满足大规模应用。2000年首个植物基因图谱拟南芥基因组通过一代测序技术破译完成,取得了植物科学研究领域里程碑式突破[2]。2005年之后,测序技术发生革命性进步,通过边合成边测序的方法,以Roche 454、Illumina Solexa/HiSeq和ABI SOLiD技术为代表的第二代测序技术(又称高通量测序)兴起,虽然第二代测序的序列存在读长较短的不足,但也难以掩盖其与第一代相比的显著优势,尤以高通量、高速率、低成本的Illumina HiSeq为代表的测序技术为代表,极大推动了基因组和转录组测序的应用与发展,成为大规模全基因组测序技术的主导。与前两代相比,Helicos Heliscope单分子测序仪和PacBio SMRT、Oxford Nanopore Technologies的GridION等纳米孔单分子第三代测序技术,超长读长、測序速率更高、测序过程无需进行PCR扩增,但配套软件平台和技术算法的商业化应用尚未成熟,测序错误率明显高于第二代。随着多种测序技术的开发和应用,加速并扩大了研究人员对植物演化及性状的认识,大量植物基因组序列被测定并取得里程碑式的研究成果。
以Sanger测序技术组装的葡萄(Vitis vinifera)基因组测序工作于2007年完成[3-4],为果树基因组测序建立了良好开端,随后Sanger测序又运用到番木瓜(Carica papaya)基因组测序中[5],Roche 454结合Sanger、Illumina测序技术代替了单一Sanger组装完成了可可(Theobroma cacao)[6]、香蕉(Musa acuminata)[7]等复杂的基因组测序,之后多种热带植物的全基因组测序工作在测序技术大发展的背景下相继完成并公布,获得了高质量的全基因组数据,为热带植物分子育种提供了优良的数据基础。本文以plaBiPD在线网站(https://www.plabipd.de/index.ep)为查询数据库,回顾并分析了包括热带水果、热带经济作物和药用植物在内的13种具代表性的热带植物的全基因组测序研究结果,探讨了各植物间全基因组测序历程和面临的难题,以及基于基因组关联转录组学挖掘的重要研究。
1 主要热带植物全基因组测序研究概况
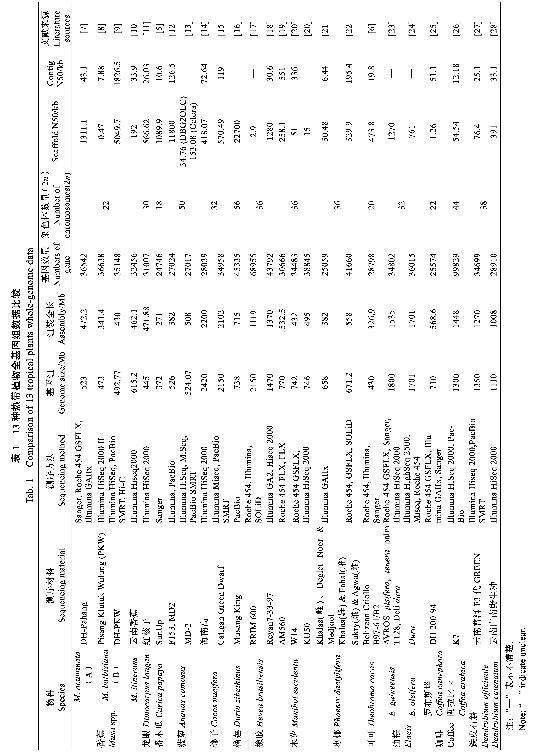
通过比较香蕉(Musa spp.)、龙眼(Dimocarpus longan)、番木瓜(Carica papaya)、菠萝(Ananas comosus)、椰子(Cocos nucifera)、榴莲(Durio zibeth inus)、橡胶(Hevea brasiliensis)、木薯(Manihot esculenta)、枣椰(Phoenix dactylifera)、可可(Theobroma cacao)、油棕(Elaeis guine ensis)、咖啡(Coffea canephora)、鐵皮石斛(Dendr obium officinale)等在内的13种热带植物的基因组信息(表1),发现最先测序组装的番木瓜基因组大小在13种植物中最小,且采用第一代Sanger测序技术。香蕉和龙眼基因组大小相当,多以第二代测序技术为主要测序方式。而较为复杂的大型乔木椰子、橡胶和菠萝基因组数据较大,均采用了第二代测序技术与第三代测序技术相结合的方法。在起步较晚的香蕉B基因组、矮种椰Catigan Green Dwarf以及云南普洱F3代GREEN均启用了第三代测序PacBio SMRT或Hi-C技术。从测序技术来看,热带植物多采用主流的第二代Illumina Hiseq 2000。从基因数目来看,番木瓜基因组注释的数量最少,咖啡(阿拉比卡)基因组注释的数量最多,达99 829。但是基因最大的椰树基因组上注释的基因数目远小于咖啡(阿拉比卡),可见基因组大小和基因数目之间并没有直接关系。
多数植物基因组由于远缘杂交、自交不亲和及基因组较大且基因组杂合度、倍性高等原因,导致基因组组装难度加大。N50指的是将所有组装的基因组序列从长到短依次排列,从最长的序列开始叠加,当叠加总长达到所有序列总长的50%时,被叠加的那条序列即为N50长度,N50越大代表序列组装质量越好。在具有高度相似序列重复和高杂合度的植物中,基因组序列分布分散且重复出现导致Scaffold N50相对较短,这使得即使对于长插入片段精准度下降,因此可能导致Scaffold延伸的破坏,N50指标下降。香蕉B基因组PKW、橡胶RRIM 600、木薯KU50、铁皮石斛云南普洱F3代GREEN和枣椰(Khalas(雌)、Deglet Noor & Medjool)的测序组装Scaffold N50和Contig N50指标均较低,其主要原因在香蕉PKW基因组上,主要是由于测序采用基因组读数映射导致组装质量欠佳,而在橡胶RRIM 600、木薯KU50和铁皮石斛云南普洱F3代GREEN则由于基因组测序覆盖度较低、测序材料不理想而致使组装难度加大。可见,为确保高质量的基因组测序,应特别注意测序材料的选择。
1.1热带水果
1.1.1 香蕉 香蕉是芭蕉科大型单子叶植物,是重要热带水果,同时也是世界第四大粮食作物。香蕉存在4种基因组,A基因组(Musa acuminata)[7]、B基因组(M.balbisiana)[8]、S基因组(M. schizo carpa)[29]和T基因组(Australimusa)。自然演化和种间杂交导致A和B基因组产生多种基因型(二倍体:AA、BB、AB;三倍体:AAA、AAB、ABB;四倍体:AAAB、AABB、ABBB),大多数可食用栽培品种是三倍体。在人为驯化过程中培育出野生二倍体杂交的三倍体单性结实杂种,通过营养繁殖进行扩繁。杂交基因组含有不确定数量的A和B基因组等位基因,使香蕉遗传性状的研究复杂化。香蕉在生产过程中面临的毁灭性威胁是来自于由尖孢镰刀菌古巴专化型(Fusar ium oxys porumf. sp.cubense,Foc)引起的枯萎病, 又称巴拿马病,且该病原菌热带型4号小种(TR4)危害最为严重,尚无有效的根治方法。因此,选育抗生物和非生物胁迫的优良香蕉品系迫在眉睫,而基因组测序为挖掘优良基因信息提供了重要的参考依据。本文主要介绍了A、B和阿宽蕉基因组。
香蕉全基因组测序在2012年首次报道,DHont等[7]利用Roche 454 GSFLX组合Sanger、Illumina GAIIx测序技术以20.5倍覆盖度读取了二倍体马来西亚小果野蕉DH-Pahang(AA)基因组序列523 Mb基因组序列草图,Illumina读数50倍覆盖度进行数据校正,最终组装了24 425个contig和7513个Scaffold,总长为472.2 Mb,覆盖90%的全基因组序列和92%的注释基因,注释蛋白编码基因36 542个,非编码RNA含819个转运RNA(tRNA)、235个微小RNA(miRNA)。基于比较基因组和转录组数据解析了芭蕉科的基因组进化特征,为后续香蕉遗传改良研究奠定了重要基础。
随后,香蕉B基因组公布。野生二倍体Pisang Klutuk Wulung(PKW)是B基因组组合三倍体的最早祖先亲本,对香蕉黑条叶斑病具一定抗性。Davey等[8]利用Illumina HiSeq 2000 II对PKW进行测序,通过将PKW gDNA读数映射到A基因组中提取共有序列和De novo组装分别得出341.4、339.3 Mb的基因组序列,注释蛋白编码基因36 638个,获得的B基因组的序列大小是A基因组的79%,与A基因组存在大量序列差异,每23.1 bp存在1个纯合单核苷酸多态性(SNP),且每55.9 bp存在1个杂合SNP,表明香蕉基因组的高度杂合。将香蕉PKW基因组读数与已报道的A基因组进行映射比对,共发现PKW重复区域占B基因组的26.85%(108.1 Mb),且A、B基因组中miRNA数量较接近,B基因组中存在一定数量的新miRNA。异源多倍体香蕉基因在所有染色体基因分布的差异性,及A和B基因组重复序列表征在分子遗传学研究中具有重要价值,谱系特异的多样化机制和基因组多样性,为后续更加深入理解种间或种内三倍体香蕉杂种代谢研究提供了宝贵的参考资源,弥补了B基因组研究的空白。但由于从头组装和映射读取使得组装质量和注释基因文库有效性较低。
近期,金志强团队Wang等[9]对B基因组进行了更高质量的组装,利用Illumina HiSeq 2000结合PacBio SMRT、Illumina NovaSeq 6000、Hi-C获得492.77 Mb的Scaffold,覆盖全基因组的95%,最终组装了430 Mb的全基因组序列。在11条染色体上,注释蛋白编码基因35 148个,86%的基因与转录组数据匹配。A、B基因组的基因家族扩张和收缩分析显示,与其他测序的基因组相比,在A和B基因组中显著扩张的基因家族分别有83和33个,相反,这些扩张的家族又在对立基因组中显著收缩。为研究多倍体香蕉中亚基因组的进化、遗传多样性和亚基因组的功能差异,进一步比较了A和B基因组,发现它们的分化发生在全基因组复制之后。而多倍体香蕉中亚基因组的功能分化主要是由A和B基因组之间染色体重排和基因缺失的结构变异等造成。三倍体粉蕉(Musa ABB group, cv Pisang Awak)在乙烯生物合成和淀粉代谢通路中的相关基因家族显著扩张,并且在同源三倍体的B亚基因组中表现出更广泛的同源基因表达优势。B基因组的高质量测序为A、B及同源多倍体香蕉的进化研究提供了新的见解,同时解析了B基因组相关功能基因在香蕉果实成熟过程的演变与调节。
野生近缘种含有栽培种的有益等位基因,是未来香蕉育种的重要基因库。有研究显示,中国亚热带地区广泛分布的香蕉野生近缘种云南香蕉(阿宽蕉)M. itinerans,是芭蕉属中最具FocTR4抗性和耐寒性的物种之一,这为香蕉育种中的抗病性和抗寒性提供了宝贵的资源[30]。2016年Wu等[10]利用Illumina Hiseq 2000对阿宽蕉进行测序,从头组装获得462.1 Mb基因组序列,覆盖75.2%的全基因组(615.2 Mb),预测蛋白编码基因32 456个,杂合率为0.25%,注释蛋白编码基因占比86.9%,非编码RNA中注释了345个miRNA、977个tRNA、278个核糖体RNA(rRNA)和299个小核RNA(snRNA)。组装后质量评估显示GC含量分布多在38.8%左右,与A基因组分布一致。同时,韦恩聚类分析发现阿宽蕉与A基因组含共有直系同源基因家族2727个,揭示两基因组具有更高的相似性。
1.1.2 龙眼 龙眼是无患子科(Sapindaceae)热带亚热带名贵特产水果,其果实风味独特、富含酚类等次生代谢物质,药用保健价值高。由于龙眼的遗传杂合性较高,常规育种手段改良其遗传特性耗时长、稳定性差。赖钟雄课题组Lin等[11]利用Illumina HiSeq 2000全基因组测序组装了“红核子”龙眼基因组序列445 Mb,雜合度为0.88%;并基于13个栽培龙眼种质登录的全基因组de novo从头测序,组装全长471.88 Mb的基因组图谱,覆盖106.4%的全基因组,配对末端测序获得273.44倍的覆盖度,注释蛋白编码基因310 07个,其中262 61个基因组成149 61个基因家族,非蛋白编码 RNA中共注释359个miRNA、212个rRNA、506个tRNA和399个snRNA。Scaffold N50和Contig N50分别为566.62 kb和26.04 kb,表明龙眼基因组组装质量高。SNP和插入缺失位点(InDel)分别为357737与23225。利用比较基因组学结合转录组分析,揭示了龙眼基因组的结构及其进化规律,同时明确了龙眼富含酚类等次生代谢物质及对植物病原体产生抗性的机制。
1.1.3 番木瓜 番木瓜是热带亚热带地区经济作物,其富含维生素A和木瓜蛋白酶,营养价值和药用价值较高。番木瓜与拟南芥同属于十字花目(Brassicales),在被子植物进化史上与拟南芥祖先相同[31]。转基因(抗环斑病毒)栽培种SunUp雌株的全基因组测序[5]是商业抗病毒转基因果树的首次测序,该基因组测序大小372 Mb,3倍测序深度覆盖75%的全基因组,组装基因组大小为271 Mb,杂合度为0.06%。注释基因数量为24 746个,已知DNA转座子的丰度相对较低(0.2%),转录因子家族和超家族数量为2438个,非蛋白编码RNA中注释tRNA和snRNA分别为388和47个,总体数量及多数家族数目均比拟南芥和其他常见热带植物少。
通过与现统计的主要热带植物的比较发现(表1),番木瓜基因组大小及基因数量最低,功能基因仅包含少量抗病相关基因,Ming等[5]认为是由多代近亲人工栽培繁殖导致番木瓜进化出特殊防御机制。进化分析结果显示,番木瓜与拟南芥在7200万年前分歧进化后,其进化分支较保守,基因谱系中基因组复制事件较少,甚至未发生。值得注意的是,尽管在大多数生物合成途径中基因数量减少,但番木瓜中预测与控制生长和繁殖相关的MADS-box家族成员的数量(205个,占基因组数量的47%)明显高于其他测序植物基因组中的水平(橡胶中在基因组仅占比12.5%[17])。番木瓜特异性基因显著扩增,对有关番木瓜淀粉积累运输、种子传播媒介的吸引、长日照适应、抗环斑病毒、纤维素与木质素合成、性别决定等基因研究具重要作用。番木瓜作为典型转基因植物,其核基因组中有3个位置与叶绿体插入、拓扑异构酶I识别位点密切相关,这对解析转基因植株中目的基因插入、表达及功能研究具重要意义。
1.1.4 菠萝 菠萝为凤梨科(Bromeliaceae)喜光植物,是世界第二大热带水果,同时属于景天酸代谢途径(CAM)植物。CAM途径是指生长在热带及亚热带干旱及半干旱地区的一些肉质植物(最早发现在景天科植物)所具有的一种光合作用固定二氧化碳的附加途径,其叶片气孔白天关闭,夜间开放。由于菠萝同高粱以及水稻等禾草植物共有一个遥远的祖先,菠萝成为禾谷类作物基因组研究的最优对照组。Ming等[5]利用F153品种、MD-2和1个野生品系杂交并进行了测序,组装了382 Mb的基因组,占基因组估计总长526 Mb的72.6%,并构建了25条染色体。通过与已测序的禾本科植物共线性分析发现,菠萝基因组少了一次古老的全基因组复制事件(WGD),即菠萝在历史上经历了2个全基因组复制而禾草植物中存在3次这种复制。研究人员重建了单子叶植物的7条原始染色体,推测出了从古到今的核型变化,在菠萝中第1次WGD使7条原始染色体变成14条,而后其中2条5号染色体转到了其他染色体上,使得染色体剩下12条。第2次基因组复制事件使12条染色体再次加倍变成24条,最后经历了一些融合和裂变,成为25条染色体。
另外,Ming等[5]对其中发生的景天酸代谢光合作用机制进行了详细的分析,首先结合转录组数据鉴定出了38个参与景天酸代谢途径的基因,并观察CAM相关的基因在夜间和白天是如何表达的,最终挖掘到了最重要的CA基因,它在菠萝里存在3个拷贝(α、β和γ)。其中βCA基因明显可以看出昼夜节律,在夜间和早晨有较高的表达量,而在下午到晚间表达量降低。在βCA基因中,找到了1个CCA1的结合位点,这个结合位点可以结合两个昼夜节律的关键基因CIRCA DIAN CLOCK ASSOCIATED 1(CCA1)和LATE ELONGATEDHYPOCOTYL(LHY)。即通过这个结合位点,可以控制CA基因的表達量,从而控制整个通路,使之与气孔的开放、闭合同步。除此之外,还有其他的有关昼夜调控的顺式作用元件都有在光合作用基因的拷贝中发现富集,说明CAM光合作用受昼夜节律基因顺式作用元件的调控[5]。这项发现为C3作物通过基因改造景天酸光合代谢创造新的抗旱节水作物奠定了基础。
2016年Redwan等[13]对重要商业菠萝品种MD-2进行了基因组测序,通过使用二代和三代2个测序平台,即PacBio长测序读取和Illumina精确短读取相结合,获得了估计基因组99.6%的覆盖率,注释27 017个蛋白质编码基因,确定了占基因组45.21%的重复序列。此外,菠萝果成熟RNASeq文库的差异表达揭示了与乙烯合成途径中相关的转录本,发现它们在参与调节菠萝非跃变型果实的成熟过程中发挥着重要作用。该项研究通过以MD-2菠萝基因组为例子,通过2种测序手段对1个复杂的杂合基因组进行测序,证明了混合技术相结合既经济又准确,为了解植物复杂生物学过程提供了更加可行的方法。
1.1.5 椰子 椰子为棕榈科(Arecaceae)乔木,是重要的热带水果及油料作物,在热带地区近百个国家广泛种植。椰子繁育周期较长,有高种、矮种和介于两者间的杂交种椰子3种生态型。高种椰子高度异交、种植范围商品性最大;矮种椰子高度自交、营养生长周期较短;中间型的杂种椰子则具有生长周期短的杂种优势。
中国热带农业科学院牵头的研究团队利用Illumina HiSeq 2000测序技术完成“海南高”椰子的全基因组测序工作,最终组装了2.20 Gb,读取深度173.32倍,覆盖90.91%的全基因组和96.78%的基因编码区,注释蛋白编码基因28 039个。基因组注释结果显示,72.75%的椰子基因组由转座因子组成,其中长末端重复序列(LTRs)反转录转座子元件占最大比例(92.23%),这一数量远高于之后的矮种椰子[14-15]。K-mer分析显示椰子是一种低杂合度、高比例重复序列的二倍体植物。在椰子中共鉴定出119个反向转运蛋白基因和67个离子通道基因,2个基因家族均发生了显著扩张,涉及与椰树耐盐胁迫有关的Na+/H+反向转运蛋白、与椰浆中脂肪酸积累相关的肉毒碱/酰基肉碱转位酶、椰汁中钾离子积累相关的钾依赖性钠-钙交换蛋白和钾通道基因。同时进化关系显示这2个基因家族的多数亚家族的数目与拟南芥相当,均能与拟南芥中的功能基因相聚集。
近期,Lantican等[15]利用Illumina Miseq组合PacBio SMRT对矮椰子品种Catigan Green Dwarf进行测序,测序深度15倍组装2.15 Gb基因组,覆盖97.6%的全基因组。注释蛋白编码基因34 985个,注释基因占总预测基因数量的85.3%,高于“海南高”高种椰(81.2%)。同时矮种椰子基因组结构高度纯合且更简单,基因组大小远低于高种椰。高种椰和矮种椰基因组中均发现大量与椰树抗逆性及生物合成相关的基因扩张。Lantican等[15]通过对棕榈科椰子、油棕[23]、枣椰[22]之间的基因组变异及共线性分析发现,3种植物的基因组大小(2.15~2.42、0.66~0.67、1.8 Gb)和染色体数目(32、32、36)差别较大,油棕基因组中与枣椰特有的Scaffolds共线的重复基因显示,油棕与枣椰的祖先为多倍体物种,在发现矮种椰与高种椰的全基因组比对高度共线的同时,还指出椰子树起源于一种常见的多倍体祖先的再二倍化。基因组中所存在的海量信息为椰子功能基因组的挖掘、农艺性状的解析及全基因组关联分析提供了参考体系。
1.1.6 榴莲 榴莲为锦葵目锦葵亚科巨型热带常绿乔木,其果实极具经济价值,是东南亚特有的热带著名水果之一,素有“水果之王”之称,因具特殊浓郁气味而闻名。新加坡研究团队Teh等[16]利用PacBio首次对Musang King榴莲基因组进行de novo组装,153倍测序深度得到183 Gb的数据量,最终组装获得榴莲基因组大小为715 Mb,K-mer分析和流式细胞仪预估基因组大小分别为738和800 Mb,杂合度为1.14%。利用CHiCAGO技术将组装成的contig连接成Scaffold,Scaffold N50为22.7 Mb,利用Hi-C技术将Scaffold挂载到染色体水平,最终将榴莲基因组组装成30条染色体,挂载率为95%。通过联合从头注释和转录组共发现榴莲含有注释基因45 335个,其中42 747个基因可以被同源或已知蛋白数据支持。GO注释到35 975个基因,多数基因富集到防御反应、果实发育、碳水化合物和脂质代谢通路中。榴莲中包含了90.3%的高保守核心蛋白,其中68.1%为单拷贝,22.2%为重复基因。独立重复基因的保守基因暗示榴莲在进化过程中经历了1次WGD。且在进化关系上,再一次证实了榴莲与棉花间的进化分歧远晚于可可的锦葵亚科进化顺序[32]。榴莲谱系中的WGD导致与榴莲挥发物相关途径的扩展和多样化,例如涉及硫处理(包括MGL)、脂质挥发物和乙烯的途径。榴莲中这些基因的上调可能与榴莲气味中的重要成分VSC的产生增加有关,从而导致榴莲气味,榴莲的复杂香气可能与榴莲果实成熟有关。该研究将基因组、转录组和代谢组相结合,揭示了榴莲特殊香气的可能分子机制,打通了结构基因组、比较基因组、功能基因组研究的链条,对后续的基因组研究,尤其是涉及物种特异的次级代谢产物功能分析具有指导意义。
1.2热带经济作物
1.2.1 橡膠 橡胶树是天然橡胶生产的主要来源,作为热带地区重要的经济作物,其产生的胶乳和橡胶木材均具有重要商业价值,在东南亚出口贸易市场占主要份额[33]。橡胶树的基因组研究起步较晚,落后于大戟科的其他物种。高通量测序结果加深了对橡胶树遗传资源的理解,而全基因组测序则弥补了在基因组非编码区信息的空白[17]。
马来西亚研究团队Rahman等[17]利用Roche 454、Illumina和SOLiD技术首次对橡胶树进行全基因组测序,13倍覆盖度最终组装了橡胶树RRIM 600的1.1 Gb基因组序列,Scaffold N50大小为2972 bp。橡胶树基因组DNA高度重复(约78%),多为长末端重复反转录转座子,这也为橡胶基因组的组装增加了难度。预测基因68 955个,KEGG注释蛋白编码基因52 825个,非编码RNA中含729个tRNA。系统发育分析表明橡胶树与木薯在进化上关系密切,二者具有相同祖先,这与橡胶树叶绿体基因组揭示的结果一致[34]。但其测序研究中序列覆盖度低,且缺乏基于fosmid或BAC等较大的插入文库,导致基因组组装欠佳。
2016年,Tang等[18]基于RRIM 600的测序,采用Illumina GA2和Hiseq 2000对中国广泛种植的橡胶栽培种Reyan7-33-97进行全基因组测序,测序深度94倍覆盖93.8%全基因组(1.46 Gb),最终组装了1.37 Gb的高质量基因组序列,Scaffold N50为1.28 Mb,注释蛋白编码基因43 792个,非编码RNA中注释了167个rRNA、591个miRNA、697个tRNA和219个snRNA。组装比对显示由于品种间的差异导致马来西亚RRIM 600与Reyan7-33-97基因组差异较大,存在25.2 Mb的未匹配序列。对另外5个品种(PR107、Reyan8-79、RRIM600、Wenchang11和Yunyan77-4)的重测序获得1.41~1.55 Gb全基因组序列,组装的84 241个转录本与基因组高度匹配,说明基因组组装质量较好;另一方面,基于SNP的系统发育显示品种之间的遗传关系与其育种历史关系密切。通过高质量基因组装配联合转录组学数据进行分析,构建了多个与胶乳生物合成相关的基因家族,尤其发现了REF/SRPP家族显著扩张,丰富了胶乳生长发育生理学及乙烯刺激胶乳生物合产机制的认识。同样,在马来西亚橡胶树基因组中,生长素基因家族成员数量较少,乙烯响应元件结合因子(ERF)相关基因数量占较大比例,这与RRIM 600中发现的乙烯促进胶乳生物合成的结果相吻合。橡胶基因组信息的揭示为改善橡胶树乳胶的高生产能力和品种遗传选育提供了良好的基础。
1.2.2 木薯 木薯为大戟科多年生灌木,是生长在非洲、美洲、亚洲热带地区的高淀粉类块根经济作物和生物能源[19]。其抗干旱、耐贫瘠、低投入、高产出的特性,使之成为三大洲超7亿人的碳水化合物主要摄入来源[20]。常规育种手段无法突破由木薯异交及广泛的热带地理分布而导致的基因组高度杂合的障碍[35]。块根含氰酸毒素而需长时间浸泡漂洗才可食用、易受细菌性和病毒性病害,以及收获后易变质等缺点制约着其种植农业的经济发展[36-38]。木薯基因组测序工作始于2003年,仅取得约700 bp的序列。与转座子相关的重复序列在木薯临近基因间散布,以及作为远缘杂交种的木薯由于具有等位基因变异、SNP和结构多态性而令每个位点的单一参考序列推导复杂化,成为木薯基因组组装中的难题。
2009年11月,由Roche 454 FLX与FLX+超长读取技术完成了近交木薯品系AM560-2的基因组测序(http://www.phytozome.net/cassava. php/),预测基因组大小为770 Mb,组装532.5 Mb的Scaffolds,覆盖70%的木薯基因组和96%的蛋白编码基因,注释蛋白编码基因30 666个,可变剪接3485个[19]。
随后在2014年,由中国热带农业科学院热带生物技术研究所的研究团队利用Illumina HiSeq 2000和Roche 454 GSFLX对野生祖先种W14和栽培种KU50的基因组序列及之前报道的AM560基因组序列进行了比较分析,W14和KU50基因组大小和测序覆盖度分别为742 Mb/58.2%、495 Mb/66.7%,组装注释蛋白编码基因数量分别为34 483和38 845[20]。基于非编码RNA在木薯野生亚种和栽培种中的分布情况,发现W14和KU50非蛋白编码RNA中较大比例为长链非编码RNA(lncRNA),分别占基因组大小的12.6%和30.1%。W14、KU50与AM560基因组非蛋白编码基因分别共注释143/126/146个miRNA、861/707/743个tRNA、337/192/237个rRNA、139/106/89个snRNA,可看出野生亚种W14基因组中注释的非编码RNA均低于栽培种。这3个基因组序列和注释的转录组的比较分析,揭示了野生和栽培木薯在自然选择过程中,基因组中与胁迫相关、光合产物运输、淀粉高效积累及氰基化合物生物合成途径基因的进化特征,阐明了木薯进化驯化及基因组功能,为后续木薯基础生物学研究及遗传育种改良提供了重要理论基础。
1.2.3 枣椰 枣椰是棕榈科(Arecaceae)的第一个被公布基因组的物种,是中东和北非地区广泛种植的木本抗旱经济作物之一,其树龄可达百年,无性繁殖,多为雌雄异株,具有重要的经济价值和历史文化意义。但由于枣椰生长周期长,且在枣椰生长早期阶段难以区分雌株和雄株而限制了枣椰育种的发展。Al-Dous等[21]利用Illumina GAIIx对Khalas雌株进行平行测序,SOAPde novo组装获得381 Mb的基因组序列,覆盖预估枣椰基因组大小(658 Mb)的60%和90%的基因,预测蛋白编码基因28 890个,杂合率为0.7%。同时对另外8个品种进行测序,利用比较基因组学分析并揭示了350万个SNP,其中有37个SNP能够用于枣椰品种区分,除此之外还确定了与枣椰性别相关的基因组区域,为枣椰性别鉴定及遗传多样性研究提供了重要的数据支撑。
此后,Bourgis等[39]利用Roche 454 (GS FLX Titanium System) 的焦磷酸测序数据对油棕和椰枣果皮进行了比较转录组学和代谢组学研究。紧接着Al-Mssallem等[22]利用Roche 454、GSFLX和SOLiD对Khalas枣椰进行了测序,获得的高通量读数覆盖枣椰预估基因组(671.2 Mb)的90.2%,最终组装获得558.02 Mb基因组序列,注释蛋白编码基因41 660个,非编码RNA中注释了414个tRNA、677个rRNA、62个snRNA。遗传多样性分析表明,枣椰抗逆性和糖代谢相关基因在SNP密度相对较低的染色体区域富集。研究揭示了枣椰基因组的倍增与进化,以及枣椰果实糖类代谢和累积的过程和机制,联合基因组和转录组数据为枣椰及棕榈科植物基因组的进一步研究奠定了重要基础。此外,该研究团队还对枣椰的产能和光合作用的细胞器(线粒体与叶绿体)基因组,以及枣椰基因模型和枣椰果实发育分析进行了重要研究。
Hazzouri等[40]以Al-Mssallem等[22]組装的Khalas基因组为参考基因组,利用Illumina HiSeq 2500对来自12个国家的62种枣椰树的基因组进行了全基因组重测序,平均测序深度为20.8倍,得出栽培枣椰中第一个完整的超过717万个高质量的SNP综合目录。利用全基因组SNP及比较基因组揭示了物种遗传和表型多样性可能机制,有望应用于枣椰重要农艺性状改良。另外确定了枣椰果实颜色多态性的等位基因,发现枣椰和油棕进化上虽有差异,但存在遗传平行性,可共享遗传信息促进二者的育种改良。
1.2.4 可可 可可是重要的热带经济作物,是巧克力的原材料,同时也是锦葵科(Malvaceae)最早完成基因组测序的物种。高品质可可市场需求高,但传统风味可可品种产量低抗病性差,改良育种成为必然需要。Argout等[6]利用Roche 454 GS FLX、Illumina GAIIx和Sanger测序技术对多代自体受精而高度纯合的Belizean Criollo基因型B97-61/B2的基因组进行了测序,产生了26 Gb原始数据,组装出25 912个Contig和4792个Scaffold,总长326.9 Mb,占可可基因组预估大小(430 Mb)的76%。注释蛋白编码基因28 798个,其中23 529个(82%)锚定在10条染色体上,非编码RNA中含83个miRNA。可可在进化过程中经历从古六倍体祖先的21条染色体进化到实际数量上的10条染色体的重组。同时发现黄酮类和萜类化合物等相关基因家族在进化过程中发生扩张,与可可风味品质及抗性关系密切,为可可改良育种提供了优良的候选基因。
1.2.5 油棕 油棕是产量最高的油料作物,被誉为“世界油王”。2016年公布的油棕基因组数据中,研究者采用了Illumina HiSeq 2500和Miseq,Roche 454等二代高通量测序技术,对东南亚重要的高产母本厚壳Dura材料进行了全基因组测序,组装出了10 971个Scaffold,长度为1.701 Gb的基因组,覆盖了94.49%的高质量油棕基因组序列草图[24]。并且对17种油棕主要组织器官进行深度转录组测序,预测了近36 105个高度可靠的油棕基因,并获得了1800万个SNP,在不同地理区域的油棕中,研究者发现它们之间存在较高的基因变异,而在东南亚的Dura和Pisifera油棕树中则存在较低的变异。并在油棕的基因组中连锁图上绘制了10 000个SNP分子标记。此外,在东南亚油棕育种群体中发现了高连锁不平衡(LD),这表明LD作图在这一重要油料作物中可能是可行的。
从Dura基因组中共鉴定出566个R基因,远远少于水稻基因组中1085个R基因数量。虽然水稻基因组的大小仅为油棕的25%,但R基因的平均Ka/Ks(1.7)远高于油棕基因组中所有基因的平均Ka/Ks(1.4),表明油棕中R基因具有很强的正向选择[24]。这些研究结果为加速遗传改良和研究重要油棕性状表型变异的机制提供了宝贵的资源。
1.2.6 咖啡 咖啡是世界上消费人群最大的饮料之一,种植面积超过1100万hm2,世界商业咖啡生产树种主要为2种,分别为异交高度杂合的二倍体罗布斯塔种(Coffea canephora)和优势种异源四倍体阿拉比卡(C. arabica)。由法国研究团队Denoeud等[25]利用Roche 454 GS FLX和Sanger以30倍覆盖度产生了710 Mb基因组序列,以Illumina GAIIx测序数据60倍覆盖度改进组装得到25 216个Contig和13 345个Scaffold,总长度为568.6 Mb,占全长(710 Mb)的80%,注释蛋白编码基因25 574个。几种特异性基因家族,如参与咖啡因生成的N-甲基转移酶(NMTs)、防御相关基因以及参与次级代谢物生物合成的生物碱和黄酮类相关基因在罗布斯塔咖啡中显著扩张。同时,咖啡中编码NMT合成咖啡因的途径与可可、茶存在差别,这些基因通过连续串联重复扩张,使得咖啡中咖啡因含量高居多种植物之首。该研究通过对罗布斯塔咖啡的基因组结构分析,确定了植物谱系中咖啡因生物合成的趋同进化,并将咖啡作为菊亚纲被子植物中基因组结构演变的参考物种,也能够帮助咖啡适应气候变化。
由于阿拉比卡咖啡基因库较小,其遗传改良进程因此而受限。鉴定能够控制咖啡因含量的相关基因,将有助于育种过程中使用分子标记有选择地进行性状基因遗传改良。为获取咖啡因含量相关的SNP,Tran等[26]从232个基因型群体中选择了具极端表型的18个基因型(咖啡因含量极高或极低)进行DNA群体测序。同时,结合137倍Illumina HiSeq 2000和6倍PacBio测序深度对阿拉比卡咖啡K7品种进行基因组测序,通过SOAPde novo组装获得76 409个Scaffold,总长度为1448 Mb,高于预估的1300 Mb。此外,超过99%的转录组序列能比对到基因组上,确定了超过89%的完整BUSCO,表明高倍性杂合的阿拉比卡咖啡基因组质量较好。以咖啡相近物种番茄基因组数据作为参考,预测注释基因99 829个(数量是二倍体罗布斯塔咖啡的4倍)。鉴定了1444个与咖啡因含量相关联的非同义SNP,进一步基于KEGG代谢通路分析,发现嘌呤代谢为最常见途径,同时发现66个与咖啡因含量相关的SNP,其中10个与参与咖啡因生物合成通路上底物转化酶相关。該研究揭示了咖啡中关于咖啡因含量性状复杂的遗传背景,为阿拉比卡咖啡遗传改良提供了优良的数据基础。
1.2.7 香荚兰 香荚兰[Vanilla fragrans(Salisb.) Ames]是一种名贵的食用香料,被誉为“香料皇后”。据2015年2月8日《光明日报》报道,福建农林大学联合国家兰科植物种质资源保护中心(深圳)宣布完成了深圳香荚兰基因组的测序。这意味着香荚兰成为第一个完成测序的兰科藤本植物,同时为揭示兰科植物起源和研究其系统演化过程及提高香荚兰的产量和品质奠定了重要基础。
1.2.8 玛卡 玛卡(Lepidium meyeniiWalp, 2n=8x=64)是十字花科草本植物,生长在秘鲁中部海拔4000~4500米的山区,被誉为“南美人参”、“秘鲁人参”,具有很好的药用和经济价值,目前在我国云南和四川也广泛种植。2016年玛卡的高质量基因组组装结果公布[41]:研究者组装了743 Mb的基因组,覆盖了估计基因组的98.93%,Contig和Scaffold N50序列的分别为81 Kb和2.4 Mb。注释了96 417个编码蛋白基因,转座元素占比47.65%。通过对玛咖与亲缘关系较近的十字花科植物的比较基因组学研究表明,玛卡基因组发生了2次WGD。玛咖基因及其家族通过WGD参与非生物胁迫反应、激素信号通路和次生代谢物生物合成。WGD导致许多重复基因被保留并且随后发生了进化,这解释了玛咖在高海拔环境下的形态和生理变化(即叶片形状变小和春化丧失)。此外,还鉴定了一些阳性选择下的重复玛卡基因具有形态适应(MYB59)和发育(GDPD5和HDA9)功能。总的来说,八倍体玛卡基因组揭示了WGD在安第斯山脉植物高海拔适应性中的重要作用[41]。
1.3药用植物
1.3.1 铁皮石斛 铁皮石斛是蘭科(Orchidaceae)石斛属名贵珍稀濒危药材,极具观赏和药用价值,其富含多糖、生物碱和氨基酸等化学成分,具有降血糖、抗氧化和抗肿瘤等药理作用[42]。近年来,有关石斛属药理成分提取工艺改进和功能基因挖掘鉴定的研究日益丰富[43-46]。基因组测序工作对深层次分析铁皮石斛性状及分子育种尤为重要。
Yan等[27]结合第二代Illumina Hiseq 2000和第三代PacBio SMRT测序技术首次组装了云南普洱人工自交的GREEN F3代铁皮石斛1.35 Gb的基因组序列,覆盖94%的全基因组和91.5%的基因编码区,Scaffold N50和Contig N50大小分别为25.1 kb和76.5 bp,注释蛋白编码基因34 699个,非蛋白编码RNA中共注释396个rRNA、545个tRNA、16个sRNA、89个snRNA和1005个miRNA。鉴定了铁皮石斛基因组中一些重要生物学特征,如多个与抗旱性及真菌共生相关基因家族的扩张、与转运相关基因(蛋白质转运、有机物质转运和细胞内蛋白质转运)进化速率加快、药用成分生物合成途径相关基因,并确定了兰花完整的花序基因集。但由于测序选材为人工自交品系,使得基因组组装复杂化。
之后,Zhang等[28]利用第二代Illumina HiSeq 2000技术对采自云南广南县同物异名的野生铁皮石斛(Dendrobium catenatum)进行全基因组测序,绘制出高质量的铁皮石斛基因图谱,最终组装1.01 Gb的基因组,覆盖93%的全基因组和97%的基因编码区,Scaffold N50和Contig N50大小分别为391 kb和33.1 kb,最终注释蛋白编码基因为28 910个,有74.9%的基因与转录组数据的匹配,非编码RNA中注释了248个rRNA、310个tRNA、144个snRNA和49个miRNA。基因组大小及基因数量均低于GREEN F3代铁皮石斛。Yan等[27]和Zhang等[28]的测序结果均显示出石斛属基因组高度杂合,两者SNP分别为5 432 657与5 758 781,杂合率分别为0.48%和0.63%。且2个基因组的测序组装结果表明石斛基因组中与抗性、多糖生物合成相关基因存在大量串联重复而显著扩张。更重要的是,铁皮石斛的测序工作能为大型复杂基因组的从头测序和组装提供具成本效益的参考。
1.3.2 辣木、丹参和三七 近几年来,云南农业大学建立的云南省生物大数据重点实验室,以现代分子生物学技术为研究手段,选取云南特色生物资源为研究对象,进行全基因组测序,完成了辣木(Moringa oleiferaLam.)[47-48]、丹参(Salvia miltiorrhiza Bge.)[49]和三七[Panax notoginseng(Burk.) F. H. Chen][50]基因组相关分析,为药材的育种、药理研究、病虫害防治等提供了重要的分子生物学依据。
2 热带植物基因组重要基因挖掘
2.1抗性(R)基因
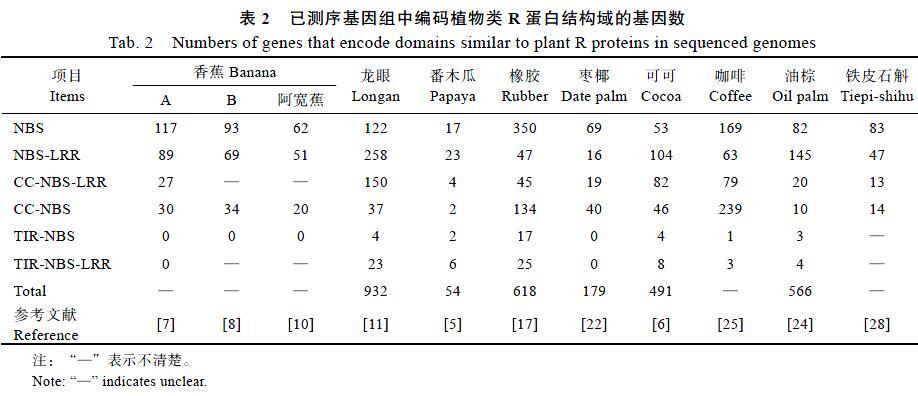
抗性(R)基因能够帮助植物产生对多种病原体和害虫的抗性,在植物病原体感知、宿主防御和细胞周期进展中起关键作用[51]。多为编码具有细胞质核苷酸结合位点-羧基末端富含亮氨酸重复序列(NBS-LRR)结构域的特定蛋白质组,NBS-LRR基因家族在植物基因组中相当丰富,约占总基因组的0.6%至约2%[51-52]。基于N-末端和C-末端结构域的结构,进一步划分为N-末端结构域或不具有CC(卷曲螺旋)/TIR(Toll-白细胞介素受体)基序、C末端结构域含或不含有LRR(富含亮氨酸的重复)基序等[53]。
基于转录组分析(表2),龙眼基因组中含有大量差异表达的植物病原体抗性基因,包括编码NBS和NBS-LRR蛋白的基因,其数量分别为122和258个,数量高于番木瓜(26/20)、铁皮石斛(83/47)、香蕉(117/89)。番木瓜中NBS型R基因数量最少[5]。香蕉基因组序列中,防御相关基因NBS基因含有117个,NBS-LRR为89个,CC-NBS-LRR为27个。对香蕉的比较基因组研究中[10],在A、B、阿宽蕉基因组中分别鉴定出117、93、62个编码NBS-基因,研究结果显示编码NBS基因数量随着各种质栖息地从潮湿热带到凉爽亚热带的过渡而减少,即与纬度上升密切相关。另一方面,单子叶植物中多不含有TIR-NBS-LRR型R基因,研究比较发现这种现象在香蕉基因组中同样存在。罗布斯塔咖啡中NBS基因存在大量基因复制,该基因家族中,42个NBS基因分布于8号染色体上,且36个NBS基因存在于基因簇中,研究推测可能是由于连锁基因家族的重复和分化进化而来[25]。
2.2次生代谢产物及活性成分相关基因
龙眼基因组中富含酚类化合物,酚类化合物主要通过莽草酸、苯丙烷类和类黄酮合成途径衍生。Lin等[11]通过比较转录组学和全基因组分析显示,龙眼基因组中3大途径结构基因显著扩张的是DHS、SDH、F3'H、ANR和UFGT等基因家族,显著较少的结构基因为PAL、CHS和F3'5'H等基因家族,這些结构基因家族均具组织特异性。其中,PAL参与木质素合成,在龙眼细胞壁中扮演重要角色,CHS参与花芽和种子中黄酮类色素合成,F3'H和F3'5'H在龙眼花色形成中起主要作用,ANR和LAR在果皮和种子中高表达、果肉中低表达表明龙眼果实的单宁组分较高。此外龙眼R2R3-MYB基因数量达94个,与拟南芥已知参与类黄酮合成的R2R3-MYB基因同源的有4个。这都为后续深入开展龙眼中次生代谢物质的研究提供了平台和机遇。黄酮类化合物参与植物多种生长发育过程,原花青素是在可可种子中大量存在的类黄酮聚合物,可可基因组中编码二氢黄酮醇-4-还原酶(DFR)及参与类黄酮生物合成途径的直系同源基因大量扩增,是可可种子中富含原花青素的重要原因[6]。
萜类化合物作为初生和次生代谢产物在植物中功能众多,除维持植物生长发育外,作为次生代谢产物在植物防御反应和信号传递过程中起重要作用[54]。铁皮石斛基因组中含有植物萜烯合成酶基因(TPS)39个,可分为7个亚家族,其中被子植物特异分支TPS-a亚家族在进化过程中经历大量串联复制而快速扩增[28]。菠萝基因组中共鉴定出5个亚家族的21个TPS成员,串联重复同样是菠萝TPS 基因重复的主要原因[55]。研究表明TPS-a亚家族成员多表达为倍半萜合成酶,在棉属植物中同样在祖先基因发生串联复制后而显著扩增[54]。而可可基因组中TPS含57个,其亚类中单萜和倍半萜数量较大,其中芳樟醇合成酶(单萜)和杜松烯合成酶(倍半萜烯)家族显著扩增,成为优良的可可昆虫抗性反应候选基因[6]。
多糖具抗氧化、增强免疫性等多种功能,其生物合成机理复杂,植物体内多糖合成与积累相关基因及其分子机制可利用基因组注释信息结合转录组学挖掘解析。铁皮石斛中存在2种主要的药用多糖,葡甘露聚糖(GM)和半乳葡甘露聚糖(GGM)。Zhang等[28]从铁皮石斛基因组发现糖基转移酶(GT)家族的13个类纤维素合成酶A(CslA)基因,该基因在铁皮石斛基因组中经历串联重复而显著扩增,认为其合成酶糖基转移与拟南芥相同来参与GM骨架合成。He等[56]基于D. offcinale基因组,利用数字基因表达谱分析鉴定出8个CslA参与甘露聚糖生物合成。
2.3胁迫响应及生长发育相关基因
基于基因组信息挖掘植物中与生长发育联系密切的基因家族成为近几年的研究热点,家族鉴定研究层出不穷。Li等[57]基于橡胶树基因组公开信息,共鉴定了81个WRKY基因,对广泛参与调节植物生长发育、抵抗生物或非生物胁迫过程的WRKY基因家族进行全基因组分析,表明HbWRKY蛋白可能参与天然橡胶生物合成的转录调控。冯新[58]基于A基因组和B基因组,对栽培蕉中与抗逆相关的SOD基因家族进行了系统鉴定,探讨了SOD在香蕉抵抗多种非生物胁迫和激素处理下调控机制。类似的研究还包括龙眼LAC家族[59]、香蕉Ran家族[60]、香蕉β-1,3葡聚糖酶基因[61]、野生蕉果皮颜色差异研究[62]和野生蕉低温响应机制研究[63]。菠萝中参与景天酸代谢光合作用中CA基因[5]、果实成熟的过程中的乙烯相关基因[13]等的挖掘为创造新的抗旱节水作物及果实的风味品质奠定了基础。木薯基因组中参与胁迫响应和生长发育的基因资源和功能研究为提高作物在胁迫、光合产物运输、淀粉高效积累等途径提供了重要的理论基础[20]。铁皮石斛基因组中一些具重要生物学特征的基因家族,如与抗旱性及真菌共生、转运相关、药用成分生物合成途径相关基因为遗传工程育种和药用成分的开发利用、规范产业发展研究提供重要资源和基础[27-28]。综上,通过植物基因组测序并从中挖掘重要农艺性状的基因,为改良作物的遗传特质提供了重要的数据支撑。
3 问题与展望
从本文统计的13种已完成全基因组测序的热带植物分析结果可以看出,虽然热带植物基因组的测序和组装均面临一定程度的困难,但各植物的基因组均有较高的组装质量。且部分植物目前已生成独立基因组数据库面向研究人员公开,如香蕉全基因组数据库(https://banana- genome- hub.southgreen.fr)、木薯全基因组数据库(https:// cas sa vagenome.org)、菠蘿基因组数据库(http:// pin e apple.angiosperms.org/pineapple/html/index. html)[64]等。基于基因组数据信息,通过生物信息学分析技术和高通量测序技术,能够深入解析物种起源,挖掘改良遗传育种和控制植物性状等相关基因,为热带植物基因组学的研究提供序列数据参考。多倍体基因组内各单倍型序列相似性较高,使得同源多倍体组装到染色体水平的难度加大。如荔枝、枇杷、火龙果和芒果等植物基因组测序还未完成,其中芒果基因组的研究目前主要集中在基因组大小测定、变异分析以及微卫星标记[65-66],未来在前列技术研究基础上有望揭示全基因组信息。第三代测序中Hi-C(染色质构象捕获)技术挂载到染色体水平能够为解决这种难题提供技术支持,利用最新的ALLHiC算法组装多倍体、高杂合等复杂基因组[67-68]。同时,对于异源多倍体基因组的组装,使用WGS和超高密度连锁作图的组合方法也能解决其组装障碍[69-70]。这都可为植物重要性状相关基因的发现、克隆、功能验证和进化分析方面的研究提供极大便利。
随着全基因组测序技术的快速发展,测序成本大大降低、测序速率显著提高,高通量测序技术应用范围更广。摆脱单一基因组研究而深入基因组关联分析研究,更重要的是,如同香蕉基因组测序对野生近缘种香蕉进行测序组装,利用日益优良的测序技术深度挖掘重要野生近缘物种,将极大促进热带或更多地区野生植物宝贵基因资源的保护和利用,壮大奥秘的植物基因资源库。
参考文献
[1]Sanger F, Air G M, Barrell B G, et al.Nucleotide sequence of bacteriophage φX174 DNA[J]. Nature, 1977, 265(5596): 687-695.
[2]The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plantArabidopsis thaliana[J]. Nature, 2000, 408(6814): 796-815.
[3]Jaillon O, Aury J M, Noel B,et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla[J]. Nature, 2007, 449(7161): 463-467.
[4]Velasco R, Zharkikh A, Troggio M,et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety[J]. PLoS One, 2007, 2(12): e1326.
[5]Ming R, Hou S, Feng Y,et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus)[J]. Nature, 2008, 452(7190): 991-996.
[6]Argout X, Salse J, Aury J,et al. The genome ofTheobroma cacao[J]. Nature Genetics, 2011, 43(2): 101-108.
[7]DHont A, Denoeud F, Aury J-M,et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants[J]. Nature, 2012, 488(7410): 213-217.
[8]Davey M W, Gudimella R, Harikrishna J A,et al. A draftMusa balbisiana genome sequence for molecular genetics in polyploid, inter- and intra-specificMusahybrids[J]. BMC Genomics, 2013, 14: 683.
[9]Wang Z, Miao H X, Liu J H, et al.Musa balbisianagenome reveals subgenome evolution and functional divergence[J]. Nature Plants, 2019, 5(8): 810-821.
[10]Wu W, Yang Y L, He W M,et al. Whole genome sequencing of a banana wild relativeMusa itineransprovides insights into lineage-specific diversification of theMusa genus[J]. Scientific Reports, 2016, 6: 31586.
[11]Lin Y L, Min J M, Lai R L,et al. Genome-wide sequencing of longan (Dimocarpus longan Lour.) provides insights into molecular basis of its polyphenol-rich characteristics[J]. GigaScience, 2017, 6(5): 1-14.
[12]Ming R, Vanburen R, Wai C M,et al. The pineapple genome and the evolution of CAM photosynthesis[J]. Nature Genetics, 2015, 47(12): 1435-1442.
[13]Redwan R M, Saidin A, Kumar S V. The draft genome of MD-2 pineapple using hybrid error correction of long reads[J]. DNA Research, 2016, 23(5): 427-439.
[14]Xiao Y, Xu P, Fan H,et al. The genome draft of coconut (Cocos nucifera)[J]. GigaScience, 2017, 6(11): 1-11.
[15]Lantican D V, Strickler S R, Canama A O,et al.De novogenome sequence assembly of dwarf coconut (Cocos nuciferaL. ‘Catigan Green Dwarf) provides insights into genomic variation between Coconut types and related palm species[J]. G3: Genes, Genomes, Genetics, 2019, 9(8): 2377-2393.
[16]Teh B T, Lim K, Young C H,et al.The draft genome of tropical fruit durian (Durio zibethinus)[J]. Nature Genetics, 2017, 49(11): 1633-1641.
[17]Rahman A Y A, Usharraj A O, Misra B B,et al. Draft genome sequence of the rubber treeHevea brasiliensis[J]. BMC Genomics, 2013, 14: 75.
[18]Tang C, Yang M, Fang Y J,et al. The rubber tree genome reveals new insights into rubber production and species adaptation[J]. Nature Plants, 2016, 2(6): 16073.
[19]Prochnik S, Marri P R, Desany B,et al. The cassava genome: Current progress, future directions[J]. Tropical Plant Biology, 2012, 5(1): 88-94.
[20]Wang W Q, Feng B X, Xiao J F,et al. Cassava genome from a wild ancestor to cultivated varieties[J]. Nature Communications, 2014, 5(1): 5110.
[21]Al-Dous E K, George B, Al-Mahmoud M E,et al.De novogenome sequencing and comparative genomics of date palm (Phoenix dactylifera)[J]. Nature Biotechnology, 2011, 29(6): 521-527.
[22]Al-Mssallem I S, Hu S, Zhang X,et al. Genome sequence of the date palmPhoenix dactyliferaL.[J]. Nature Communications. 2013, 4(1): 2274.
[23]Singh R, Ong-Abdullah M, Low E L,et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds[J]. Nature, 2013, 500(7462): 335-339.
[24]Jin J, Lee M, Bai B,et al. Draft genome sequence of an eliteDurapalm and whole-genome patterns of DNA variation in oil palm[J]. DNA Research, 2016, 23(6): 527-533.
[25]Denoeud F, Carretero-Paulet L, Dereeper A,et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis[J]. Science, 2014, 345(6201): 1181-1184.
[26]Tran H T M, Ramaraj T, Furtado A,et al. Use of a draft genome of coffee (Coffea arabica) to identify SNPs associated with caffeine content[J]. Plant Biotechnology Journal, 2018, 16(10): 1756-1766.
[27]Yan L, Wang X, Liu H,et al.The Genome ofDendrobium officinaleilluminates the biology of the important traditional Chinese orchid herb[J]. Molecular Plant, 2015, 8(6): 922-934.
[28]Zhang G Q, Xu Q, Bian C,et al. TheDendrobium catenatumLindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution[J]. Scientific Reports, 2016, 6: 19029.
[29]Belser C, Istace B, Denis E,et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps[J]. Nature Plants, 2018, 4(11): 879-887.
[30]Li W M, Dita M, Wu W,et al. Resistance sources toFusarium oxysporumf. sp.cubensetropical race 4 in banana wild relatives[J]. Plant Pathology, 2015, 64(5): 1061-1067.
[31]Wikstr?m N, Savolainen V, Chase M W. Evolution of the angiosperms: calibrating the family tree[J]. Proceedings of the Royal Society of London. Series B: Biological Sciences, 2001, 268(1482): 2211-2220.
[32]Alverson W S, Whitlock B A, Nyffeler R,et al.Phylogeny of the core Malvales: evidence fromndhFsequence data[J]. American Journal of Botany, 1999, 86(10): 1474-1486.
[33]Prabhakaran Nair K P. The agronomy and economy of important tree crops of the developing world[M]. Burlington: Elsevier, 2010.
[34]Tangphatsornruang S, Uthaipaisanwong P, Sangsrakru D,et al. Characterization of the complete chloroplast genome ofHevea brasiliensisreveals genome rearrangement, RNA editing sites and phylogenetic relationships[J]. Gene, 2011, 475(2):104-112.
[35]De Carvalho R, Guerra M. Cytogenetics ofManihot esculentaCrantz (cassava) and eight related species[J]. Hereditas. 2002, 136(2): 159-168.
[36]Boher B, Verdier V. Cassava bacterial blight in Africa: the state of knowledge and implications for designing control strategies[J]. African Crop Science Journal, 1994, 2(4): 505-509.
[37]Reilly K, Bernal D, Cortés D F,et al. Towards identifying the full set of genes expressed during cassava post-harvest physiological deterioration[J]. Plant Molecular Biology, 2007, 64(1-2): 187-203.
[38]Patil B L, Fauquet C M. Cassava mosaic geminiviruses: actual knowledge and perspectives[J]. Molecular Plant Pathology. 2009, 10(5): 685-701.
[39]Bourgis F, Kilaru A, Cao X,et al. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(44): 12527-12532.
[40] Hazzouri K M, Flowers J M, Visser H J, et al. Whole genomere-sequencing of date palms yields insights into diversificationof a fruit tree crop[J]. Nature Communications,2015, 6: 8824.
[41] Zhang J, Tian Y, Yan L, et al. Genome of plant maca(Lepidium meyenii) illuminates genomic basis for high- altitudeadaptation in the central Andes[J]. Molecular Plant,2016, 9(7): 1066-1077.
[42] 孫 恒, 胡 强, 金 航, 等. 铁皮石斛化学成分及药理活性研究进展[J]. 中国实验方剂学杂志, 2017, 23(11):225-234.
[43] 王丛巧, 王培育, 郭艳芳, 等. 昼夜温差处理下铁皮石斛原球茎松柏苷和紫丁香苷含量的测定[J]. 热带作物学报,2019, 40(2): 261-268.
[44] 林小苹, 赖钟雄. 不同光质条件下铁皮石斛多糖含量与磷酸烯醇式丙酮酸羧化酶基因表达变化[J]. 热带作物学报,2017, 38(5): 838-842.
[45] 黄晓君, 聂少平, 王玉婷, 等. 铁皮石斛多糖提取工艺优化及其成分分析[J]. 食品科学, 2013, 34(22): 21-26.
[46] 蔡璨璨, 李 卿, 段承俐, 等. 铁皮石斛Csl 基因家族生物信息学及表达分析[J]. 基因组学与应用生物学, 2019,38(5): 2159-2166.
[47] Tian Y, Zeng Y, Zhang J, et al. High quality reference genomeof drumstick tree (Moringa oleifera Lam.), a potentialperennial crop[J]. Science China Life Sciences, 2015, 58(7):627-638.
[48] Chang Y, Liu H, Liu M, et al. The draft genomes of fiveagriculturally important African orphan crops[J/OL]. GigaScience,2019, 8(3). https://doi.org/10.1093/gigascience/giy152.
[49] Zhang G, Tian Y, Zhang J, et al. Hybrid de novo genomeassembly of the Chinese herbal plant danshen (Salviamiltiorrhiza Bunge)[J]. GigaScience, 2015, 4: 62.
[50] Chen W, Kui L, Zhang G, et al. Whole-genome sequencingand analysis of the Chinese herbal plant Panax notoginseng[J]. Molecular Plant, 2017, 10(6): 899-902.
[51] DeYoung B J, Innes R W. Plant NBS-LRR proteins inpathogen sensing and host defense[J]. Nature Immunology,2006, 7(12): 1243.
[52] Mun J H, Yu H J, Park S, et al. Genome-wide identificationof NBS-encoding resistance genes in Brassica rapa[J]. MolecularGenetics & Genomics, 2009, 282: 617-631.
[53] McHale L, Tan X, Koehl P, et al. Plant NBS-LRR proteins:adaptable guards[J]. Genome Biology, 2006, 7(4): 212.
[54] 李 威. 雷蒙德氏棉和亞洲棉萜类化合物合成关键基因家族的全基因组鉴定和系统发育研究[D]. 杭州: 浙江大学, 2014.
[55] Chen X, Yang W, Zhang L, et al. Genome-wide identification,functional and evolutionary analysis of terpene synthasesin pineapple[J]. Computational Biology and Chemistry,2017, 70: 40-48.
[56] He C, Zhang J, Liu X, et al. Identification of genes involvedin biosynthesis of mannan polysaccharides in Dendrobiumofficinale by RNA-seq analysis[J]. Plant Molecular Biology,2015, 88(3): 219-231.
[57] Li H L, Guo D, Yang Z P, et al. Genome-wide identificationand characterization of WRKY gene family in Hevea brasiliensis[J]. Genomics, 2014, 104(1): 14-23.
[58] 冯 新. 香蕉SOD 基因家族的全基因组鉴定及功能分析[D]. 福州: 福建农林大学, 2016.
[59] 徐小萍, 陈晓慧, 吕科良, 等. 龙眼漆酶家族成员全基因组结构与功能分析[J]. 应用与环境生物学报, 2018, 24(4):833-844.
[60] 张雅玲, 方智振, 赖钟雄. 香蕉Ran 家族基因的全基因组分析[J]. 江西农业大学学报, 2015, 37(1): 157-162.
[61] 陈芳兰. 野生蕉β-1,3 葡聚糖酶基因克隆及抗寒相关功能分析[D]. 福州: 福建农林大学, 2016.
[62] 邓素芳. 基于RNA-Seq 的野生蕉(Musa itinerans)果皮颜色差异形成的分子机制研究[D]. 福州: 福建农林大学,2018.
[63] 刘炜婳. 基于全转录组学的野生蕉(Musa itinerans)低温胁迫响应机制研究[D]. 福州: 福建农林大学, 2018.
[64] Xu H M, Yu Q Y, Shi Y, et al. PGD: Pineapple genomicsdatabase[J]. Horticulture Research, 2018, 5: 66.
[65] 柳 觐, 李开雄, 孔广红, 等. 云南芒果种质基因组大小测定与变异分析[J]. 热带亚热带植物学报, 2015, 23(4):386-390.
[66] Ravishankar K V, Dinesh M R, Nischita P, et al. Developmentand characterization of microsatellite markers in mango(Mangifera indica) using next-generation sequencing technologyand their transferability across species[J]. MolecularBreeding, 2015, 35(3): 93.
[67] Zhang J, Zhang X, Tang H, et al. Allele-defined genome ofthe autopolyploid sugarcane Saccharum spontaneum L.[J].Nature Genetics, 2018, 50(11): 1565-1573.
[68] Zhang X, Zhang S, Zhao Q, et al. Assembly of allele-aware,chromosomal-scale autopolyploid genomes based on Hi-Cdata[J]. Nature Plants, 2019, 5(8): 833-845.
[69] Chapman J A, Mascher M, Buluc A, et al. A whole-genomeshotgun approach for assembling and anchoring the hexaploidbread wheat genome[J]. Genome Biology, 2015, 16: 26.
[70] Ming R, Man Wai C. Assembling allopolyploid genomes: nolonger formidable[J]. Genome Biology, 2015, 16: 27.
