利用CRISPR/Cas9系统对人A549肺癌细胞NRF2基因的稳定敲除及其功能研究
2019-12-13龚美玲张琳琳郑翠侠
龚美玲,张琳琳,郑翠侠
同济大学附属杨浦医院呼吸内科,上海200090
核转录因子E2相关因子2[nuclear factor(erythroid-derived 2)-like 2,NRF2]是一个亮氨酸拉链转录因子,与核转录因子E2(nuclear factor-erythroid derived 2,NF-E2)、核转录因子E2相关因子1(nuclear factor erythroid 2-related factor 1,NRF1)及核转录因子E2相关因子3(nuclear factor erythroid 2-related factor 3,NRF3)等同属Cap“n”Collar(CNC)家族[1-2]。NRF2是一个抵抗氧化应激的关键核转录因子[3]。在正常稳态环境下,NRF2与细胞质中Kelch样环氧氯丙烷相关蛋白1(Kelch-like ECH-associated protein 1,Keap1)结合,导致NRF2被降解。而在氧化应激环境中,KEAP1与NRF2解离,NRF2进入细胞核中与小Maf蛋白形成二聚体结构与抗氧化元件ARE结合,激活下游抗氧化基因、解毒基因及细胞代谢相关基因的表达[4]。NRF2已被证明与多种肿瘤发生、发展及耐药相关,如乳腺癌、结肠癌、皮肤肿瘤、膀胱癌及肺癌等[5-10]。
NRF2拥有7个高度保守的Neh(Nrf2-ECH homology)同源结构域,即Neh1~Neh7。其中Neh1位于近羧基端,主要包含一个碱性亮氨酸拉链bzip(basic leucine zipper)区,是一个DNA结合域,而且可以与小Maf蛋白形成二聚体促进与抗氧化元件ARE结合[11-13];Neh2包含7个可受泛素化修饰的赖氨酸残基以及2个Keap1结合位点(ETGE和DLG),对NRF2的活性和稳定性的调节极为重要[14-15];Neh3区域通过招募共激动剂染色质解螺旋酶DNA结合蛋白6(chromodomain helicase DNA binding protein 6,CHD6),上调NRF2目标基因表达水平[16];Neh4和Neh5区域协同招募CBP参与ARE调控下游靶基因[17];Neh6结构域是富含丝氨酸的区域,其参与NRF2稳定性的负调节,而且与Keap1无关[18];Neh7结构域研究较少,与视黄酸X受体α结合可抑制NRF2基因转录[19]。
本研究通过CRISPR/Cas9基因编辑技术对NRF2基因进行敲除,敲除序列选择在CNCbzip区,bzip区主要通过与小分子肌腱纤维肉瘤(small masculoaponeurotic fibrosarcoma,sMaf)蛋白形成二聚体从而与下游抗氧化元件ARE结合,促进下游靶基因的转录。该结构域一旦被敲除,NRF2主要功能丧失,下游相关抗氧化基因、解毒酶基因等表达下降,从而使肺癌细胞的增殖、迁移、侵袭能力下降。
1 材料和方法
1.1 材料
肺腺癌细胞株(A549)、人胚肾细胞株(HEK-293T)购自中国科学院典藏培养物保藏委员会细胞库/中国科学院上海生命科学研究院细胞资源中心,pLenti CRISPR v2载体购自领恪(上海)生物科技有限公司,BbsI酶、BsmBI酶、T4DNA连接酶购自美国Thermo Fisher公司,质粒抽提试剂盒购自德国Qiagen公司,DNA提取试剂盒购自生工生物工程(上海)股份有限公司,Blasticidins购自美国Invitrogen公司,LipofectamineTM2000试剂、Ham's F-12K(Kaighm's)培养基、高糖DMEM培养基、胎牛血清和胰酶均购自美国Gibco公司,实时荧光定量聚合酶链反应(real-time fluorescence quantitative polymerase chain reaction,RTFQPCR)检测试剂购自宝生物工程(大连)有限公司,结晶紫购自美国Sigma公司,细胞计数试剂盒(cell counting kit-8,CCK-8)试剂盒上海碧云天生物技术有限公司,Transwell板(24孔、孔径8.0 μm)购自美国Corning公司,寡核苷酸序列由铂尚生物技术(上海)有限公司合成。
1.2 方法
1.2.1 寡核苷酸序列合成
采用CRISPR在线设计工具(http://crispr.mit.edu/)根据评分系统,在NRF2基因的第5外显子设计2个sgRNA(sgRNA-F,sgRNA-R)。寡核苷酸序列见表1。

表1 sgRNA寡核苷酸序列Table 1 sgRNA oligonucleotide sequences
1.2.2 质粒构建[20]
把2条sgRNA寡核苷酸单链退火连接,用BsmBI酶将pLenti CRISPR v2载体线性化,然后利用T4连接酶过夜将退火后的产物连接到线性化的载体上,转化至DH5α感受态细胞中。挑单克隆菌落并抽取质粒送至公司测序验证。
1.2.3 慢病毒质粒包装[21]
病毒包装是我们实验室比较成熟的一项技术。根据pLenti CRISPR v2-NRF2 sgRNA-F/R质粒与慢病毒包装载体PSPAX2,PMD2.G之间的摩尔质量比,计算出各自需要的量,实现pLenti CRISPR v2-NRF2 sgRNA-F/R质粒的共同转染至HEK-293T细胞,6 h后换成含血清的高糖DMEM培液,48 h后收集病毒上清液,与polybreen(聚凝胺,转染增强剂)(8 μg/mL)比例充分混合后加入A549细胞培养板中,72 h后弃去培液,加入含Blasticidins(20 μg/mL)的F-12K培养基,筛选7 d后,将剩余存活细胞铺至96孔板,保证每孔1个细胞,数天后开始挑取单克隆细胞,提取细胞DNA。然后在靶序列两端设计引物(表2),RTFQ-PCR扩增产物送铂尚生物技术(上海)有限公司测序。

表2 靶序列扩增引物Tab.2 Target sequence amplification primers
1.2.4 蛋白质印迹法(Western blot)检测
采用上海碧云天生物技术有限公司的细胞裂解液提取3株纯合敲除细胞蛋白,BCA法测定蛋白浓度,按每组20 μg等量上样,转膜条件为300 mA,2 h。剪膜。采用5%BSA封闭2 h后按1∶2 000温育NRF2一抗抗体(美国Abcam公司),HO-1一抗抗体(CST)。β-actin(CST)。4 ℃慢摇过夜,12~18 h后回收一抗,1×TBST洗3次,每次10 min,室温按1∶5 000温育二抗抗体2 h,回收二抗,1×TBST洗3次,每次10 min,最后显影。
1.2.5 RTFQ-PCR检测
TRIzol法提取3株纯合敲除细胞RNA,采用宝生物工程(大连)有限公司生产的反转录及RTFQ-PCR试剂盒,比较未敲除组与敲除组NRF2基因及下游靶基因的转录水平的表达。每个样本3个复孔,分别进行3次独立重复实验。
1.2.6 平板克隆形成实验[21]
胰酶将细胞消化下来后,使用细胞计数仪计数,计3次,每孔按1 000个细胞数量进行铺板,挑选一株敲除细胞(NRF2 sgRNA-2)与未敲除细胞(A549)进行对比,10%FBS的完全培养基,在37 ℃、CO2体积分数为5%的培养箱中培养10~14 d。镜下观察到大于50个细胞的克隆出现时,终止培养。弃去培养液,PBS洗3遍,加入甲醇固定15 min,PBS洗3遍。每孔加入1 mL结晶紫10 min,吸出后缓慢冲洗干净染色液,干燥后拍照;或在显微镜下,计数每孔大于50个细胞的克隆数,计算克隆形成率;克隆形成率=克隆形成数/接种细胞数×100%。
1.2.7 细胞划痕实验[21]
将A549细胞与NRF2 sgRNA-2敲除细胞铺至6孔板,6孔板在加入培养液之前先用Mark笔在板下划3~5条等距直线,待次日细胞数量达到95%时,取出6孔板,倒掉培液,选择10 μL的移液器吸嘴用直尺比着开始划痕,每个孔里可以多划几道伤痕,PBS洗3遍,加入无血清培养基进行培养,分别在0、24和48 h时在倒置显微镜下进行定点拍照即可。
1.2.8 细胞Transwell实验[21]
将200 μL无血清细胞悬液(含3×104个细胞)接种到上室,在下室加入含10%FBS的培液、37 ℃、CO2体积分数为5%的培养箱中培养24 h,取出小室,用棉签将上室细胞擦拭干净,预冷甲醇液固定10 min,弃掉甲醇PBS洗2遍,结晶紫染色20 min,缓慢冲洗干净染色液,干燥后显微镜下拍照即可。
1.2.9 CCK-8检测[22]
将处于对数生长期的细胞铺至96孔板,每孔铺5×103个细胞,每组细胞3个复孔,待细胞完全贴壁后,每100 μL培液中加入10 μL的CCK-8试剂,放置培养箱中温育3 h,酶标仪下检测吸光值(D)值,弃去CCK-8换入正常培养液继续培养养,以此类推,分别检测在24、48和72 h的D值即可。
2 结果
2.1 重组质粒测序结果
将sgRNA退火连接到载体pLenti CRISPR v2上,转化后挑单克隆抽质粒,然后送至铂尚生物技术(上海)有限公司测序验证靶序列正确插入载体,黑色方框框住的序列即为靶序列(图1)。
2.2 NRF2纯合子敲除细胞株
将pLenti CRISPR v2-NRF2 sgRNA-F/R两个质粒通过包装慢病毒的方式共同转染入A549细胞株中,有限稀释法挑选单克隆细胞株,提取DNA后PCR扩增目的片段,最后送至铂尚生物技术(上海)有限公司测序验证敲除,根据测序结果挑选纯合子,本次实验挑选出了3株纯合敲除细胞,分别命名为NRF2 sgRNA-1、NRF2 sgRNA-2和NRF2 sgRNA-3。测序结果显示3种不同的编辑方式的确在靶序列部位部分敲除(图2)。
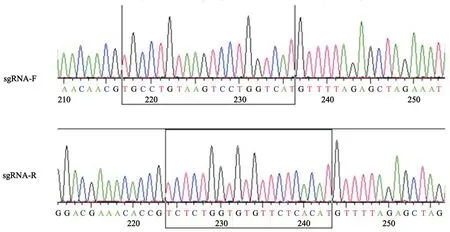
图1 靶序列插入质粒测序图Fig.1 Target sequences on plasmid sequencing maps
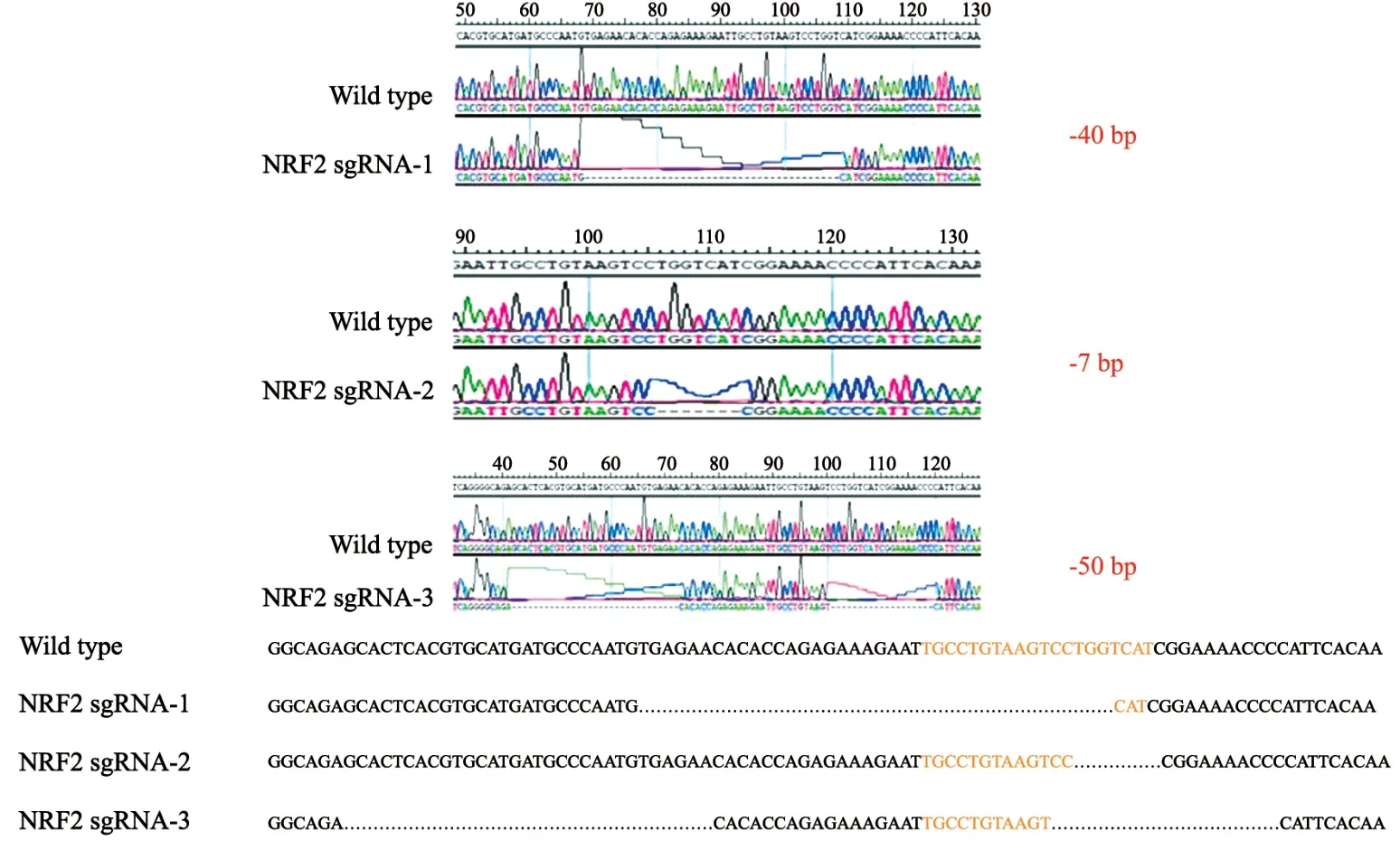
图2 3株纯合子敲除细胞株基因编辑方式Fig.2 Gene editing methods for 3 homozygous knockout cell lines
2.3 NRF2基因及其下游HO-1蛋白水平
3株纯合子敲除细胞进行NRF2蛋白表达验证,结果显示NRF2基因确被敲除。与A549细胞相比较,NRF2及其下游靶基因HO-1(血红素加氧酶-1)抗氧化酶的蛋白水平显著降低,进一步验证NRF2敲除成功(图3)。
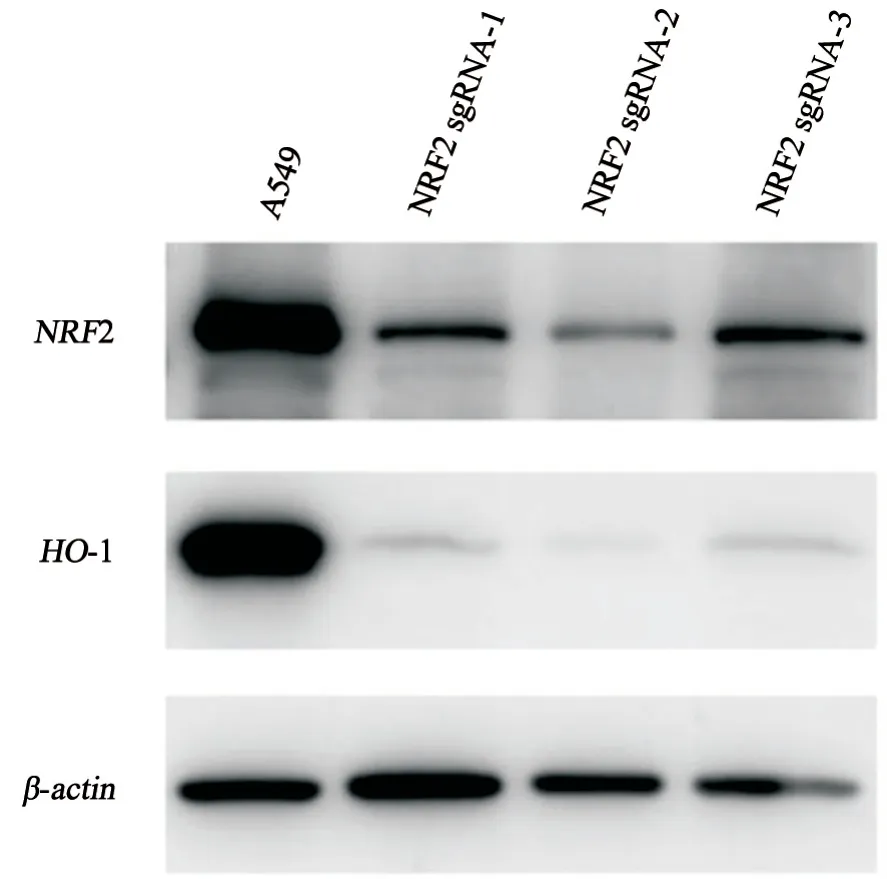
图3 3株NRF2纯合敲除的A549细胞蛋白水平结果显示NRF2及其下游靶基因HO-1表达显著降低Fig.3 The results showed that the expressions of NRF2 and its downstream target gene HO-1 were significantly decreased in 3 NRF2 homozygous knockout A549 cells
2.4 NRF2及其下游靶基因mRNA表达水平
与A549细胞相比较,NRF2敲除后其自身及下游靶基因HMOX1、HO-1、GCLC、NQO1、FTH1 mRNA表达都明显下降,说明bzip区域的敲除确实可以阻断NRF2与ARE的结合,从而抑制下游靶基因的表达,统计方法采用未配对t检验(图4)。
2.5 NRF2敲除细胞系增殖能力
通过平板克隆形成实验,可以观察到与A549细胞相比较,NRF2敲除细胞增殖能力明显低于未敲除细胞,表明NRF2在肺癌发展中起重要作用(图5)。
2.6 NRF2敲除细胞系增殖能力
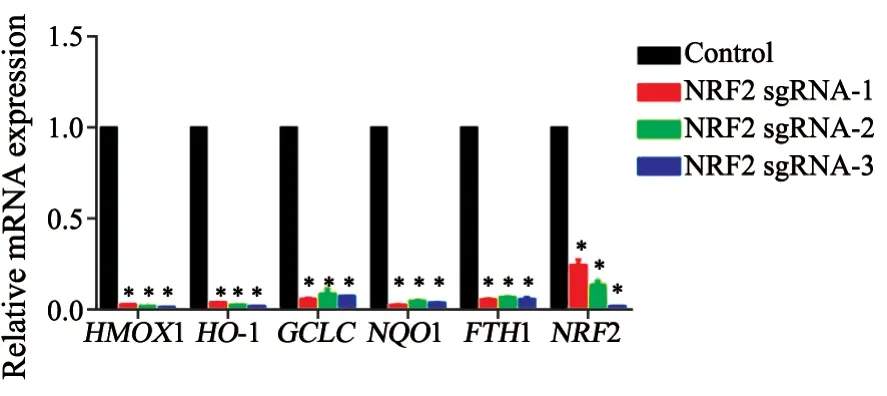
图4 NRF2敲除细胞株与A549细胞株之间NRF2下游靶基因表达的比较Fig.4 Comparison of NRF2 downstream target gene expression between NRF2 knockout cell line and A549 cell line
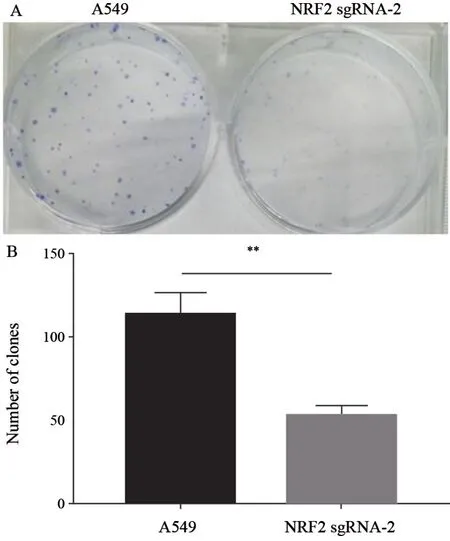
图5 A549与NRF2 sgRNA-2细胞平板克隆形成实验Fig.5 Comparison of the ability of A549 to differentiate into NRF2 sgRNA-2 cell plate clones
通过CCK-8实验方法检测活细胞数及增殖能力,可以观察到随着时间增长,NRF2敲除细胞的D值明显低于未敲除细胞,D值的大小与活细胞数成正比(图6)。
2.7 NRF2敲除细胞系迁移能力
通过细胞划痕实验可以观察到随着时间的延长,NRF2敲除细胞系伤痕愈合即迁移能力明显低于A549细胞,充分证明A549细胞NRF2基因敲除后会明显降低肿瘤细胞的迁移能力(图7)。
2.8 NRF2敲除细胞系侵袭能力
采用Transwell法比较细胞的侵袭能力,结果显示,NRF2敲除后A549细胞的侵袭能力较亲本A549细胞显著减低,表明NRF2基因敲除可以使A549肺癌细胞的侵袭能力下降(图8)。
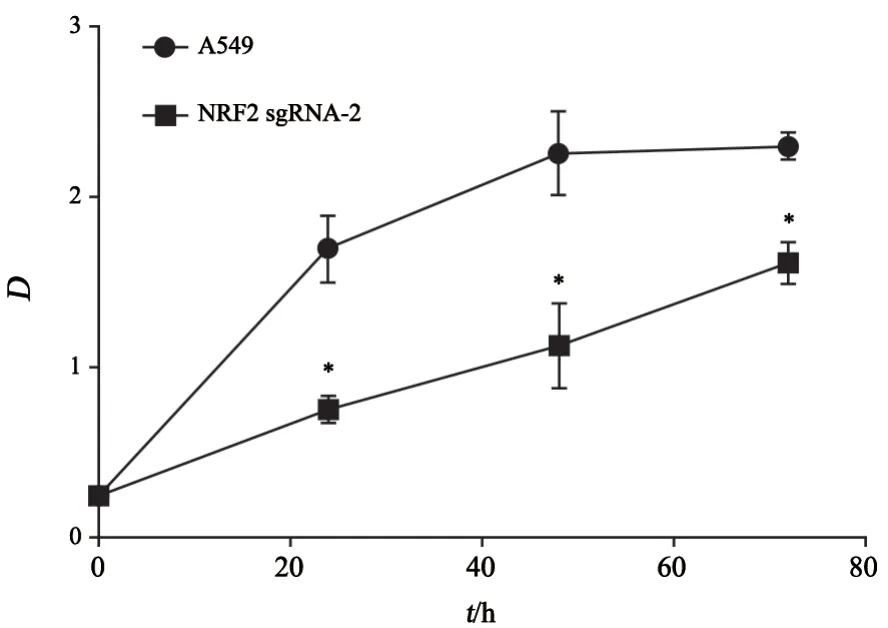
图6 A549、NRF2 sgRNA-2细胞加入CCK-8试剂后0、24、48和72 h的D值折线图Fig.6 Line diagram of D values at 0,24,48 and 72 h after CCK-8 reagent was added to A549 and NRF2 sgRNA-2 cells

图7 A549与NRF2 sgRNA-2细胞株迁移能力的比较Fig.7 Comparison of migration ability between A549 and NRF2 sgRNA-2 cell lines
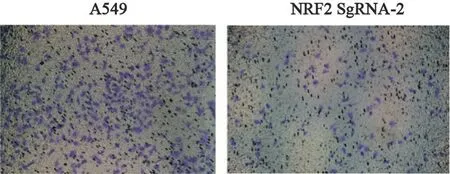
图8 A549与NRF2 sgRNA-2细胞之间侵袭能力的比较Fig.8 Comparison of invasive ability between A549 and NRF2 sgRNA-2 cells
3 讨 论
近年来,CRISPR/Cas9成为科研领域一项很热门的技术,具有设计简单、操作方便,成本低、效率高且可同时进行多位点编辑等优势。CRISPR/Cas9系统可以快速方便地在哺乳动物细胞系、动物模型中实现基因组编辑[23-25]。除此之外,该基因编辑技术在医学领域疾病治疗方面也有所进展[26-27]。此外,本次实验利用慢病毒表达载体,具有转染效率高、目的基因稳定表达等优点,可为后续其他功能研究带来方便。
NRF2基因已被证明在很多肿瘤中携带有突变或高表达状态,NRF2的表达和活性增强与肿瘤的发生、发展、耐药及预后等相关[28]。本实验选取的人A549肺癌细胞本身携带有KEAP1基因G333C突变,且NRF2基因高表达[29]。我们选择利用CRISPR/Cas9技术对NRF2基因实现稳定敲除。在NRF2基因7个Neh高度保守序列中,我们选择针对Neh1区域的CNC-bzip结构上下游设计一对sgRNA。Neh1区域包括CNC-bZIP区域,该区域是DNA结合和NRF2与sMaf蛋白结合形成二聚体所必需的区域[30]。故该结构被敲除后,NRF2与下游靶基因的抗氧化元件ARE结合能力显著降低,导致各类抗氧化基因、药物解毒相关基因等蛋白水平及mRNA表达也受到抑制。有研究报道,A549肺癌细胞NRF2基因敲除在体外移植瘤实验中单独表型即表现为增殖减慢,而且联合卡铂、顺铂等化疗药物能更明显抑制肿瘤的生长[20]。与我们体外功能研究结果相一致,克隆形成、CCK-8、细胞划痕、Transwell等实验也证明NRF2基因敲除后细胞的增殖能力、迁移能力、侵袭能力都显著降低。
利用CRISPR/Cas9慢病毒系统获得高效永久NRF2基因敲除肺癌细胞系,且NRF2基因敲除后A549肺癌细胞的增殖、迁移、侵袭能力都显著降低。
