青黛药材中非法染色剂的鉴别和亮蓝的含量测定
2019-12-05卞艳芳周春娜
宫 宇,卞艳芳,周春娜,许 琛
(海军军医大学第一附属医院/上海长海医院药学部,上海 200433)
青黛为爵床科植物马蓝[Baphicacanthuscusia(Nees)Bremek.]、蓼科植物蓼蓝(PolygonumtinctoriumAit.)或十字花科植物松蓝(IsatisindigoticaFort.)的叶或茎叶加工制得的干燥粉末、团块或颗粒,具有清热解毒、凉血消斑和泻火定惊的作用,临床主要用于口舌生疮、口腔溃疡及由于温热引起的上火症状[1-3]。现代药理研究结果表明,青黛的主要活性成分为靛蓝和靛玉红[4-6]。靛蓝主要用于纺织品的染料,同时也可以用于食品、医药和化妆品的着色;靛玉红具有凉血解毒、抗炎及抗肿瘤的作用,临床用于治疗慢性粒细胞白血病等[7]。目前对青黛的研究主要有化学成分[8]、微量元素[9]、指纹图谱[10-11]、抗真菌成分[12]和染色研究[13-14]等,由于青黛制备工艺复杂且产量低,近年来有报道不少不良商家为了提高青黛药材的外观色泽,在青黛中添加一些人工合成色素,有孔雀石绿、结晶紫和亮蓝[15]等。亮蓝是一种非偶氮类的水溶性着色剂,具有成品低廉、色泽亮丽和着色力强的特点,主要用于食品中饮料、糖果和糕点上的彩妆,《食用添加剂使用卫生标准》GB 2760-1996对其使用进行了限量规定[16]。由于亮蓝是由煤焦油中的苯胺为原料经过一系列复杂的工艺合成而来,因此具有一定的毒性。本研究根据文献报道[17-18],采用薄层色谱法(thin-layer chromatography,TLC)和超快速液相色谱-串联质谱(ultra-fast liquid chromatography coupled with triple quadrupole mass spectrometer,UFLC-MS/MS)法进行鉴别与定量。对10批不同来源的青黛进行了染色鉴别,并对鉴别出的亮蓝进行了含量测定研究,为青黛药材中的非法染色鉴别提供依据,从而保证临床用药安全。
1 材料
1.1 仪器
Shimadzu UFLC-8040型超高效液相串联质谱仪(日本岛津仪器公司),包括二元高压洗脱泵,UV检测器;Labsolution工作站,BSA-124S型电子天平(德国赛多利斯),TLC硅胶G薄层色谱板(美国Merck公司)。
1.2 药品与试剂
青黛药材分别来源于福建、云南和江苏等地区,所有药材购自上海凯旋门中药材市场和上海长海医院中药房,所有药材均经卞艳芳主管药师鉴定为爵床科植物马蓝的叶或茎叶加工制得的干燥粉末,有2批样品疑似染色。对照品:亮蓝(批号:GBW10004,质量分数97.3%)购自国家标准物质中心;亮蓝G、酸性绿50、亚甲基蓝和甲基蓝均购自Dr.Ehrenstorfer GmbH公司。乙腈为色谱纯(美国Fisher公司);水为超纯水(美国Milli-Q公司);甲酸为色谱纯;乙酸乙酯、无水乙醇、正丁醇、氢氧化铵及乙醇等均为分析纯(上海国药试剂化学有限公司)。
2 方法与结果
2.1 TLC鉴别
取上述取10个不同批号产地的青黛药材粉末,称取0.5 g,加乙醇25 ml,超声处理10 min,滤过,取续滤液作为供试品溶液。分别精密称取亮蓝、亮蓝G、酸性绿50、亚甲基蓝和甲基蓝对照品适量,加甲醇稀释制成每1 ml分别含1 mg的溶液,作为对照品溶液。精密吸取上述供试品和对照品溶液各2 μl,按照TLC法(通则0502)试验,分别点于同一硅胶G薄层板上,以乙酸乙酯-正丁醇-乙醇-氨水-水(V∶V∶V∶V∶V=1∶3∶3∶1∶1)为展开剂,展开8 cm,取出,晾干,在日光下检视。供试品色谱中,只有3号和8号供试品疑似在与亮蓝对照品位置上有相同颜色的斑点,其他供试品未有与上述对照品色谱相应的位置上显相同颜色的斑点,见图1。
2.2 液相色谱和质谱条件
色谱条件:UFLC-8040超高效液相串联质谱仪(日本岛津仪器公司),Shim-packVP-ODS色谱柱(2.1 mm×100 mm,2 μm),流动相为乙腈-0.01%甲酸水(V∶V=36∶64),流速0.4 ml/min;柱温40 ℃,进样体积1 μl。质谱条件为电喷雾ESI离子源,正离子扫描,干燥气和雾化器为液氮,加热器为空气,雾化器流量3 L/min,干燥器流量10 L/min,加热器流量10 L/min,接口温度300 ℃,脱溶剂管温度250 ℃,质量扫描范围50~1 000;驻留时间100 msec,多反应监测(MRM)模式,亮蓝在MRM检测模式下,质谱参数为:一级离子(m/z)793.2,二级离子(m/z)为623.1 CE能量为-45.0 eV,713.1 CE能量为-39.0 eV,543.3 CE能量为-50.0 eV,m/z 623.1为定量离子,一级质谱和二级质谱图见图2。
2.3 对照品溶液的制备
精密称取亮蓝对照品5.16 mg置于25 ml容量瓶中,加乙醇稀释至刻度,摇匀,作为对照品储备溶液。精密吸取上述亮蓝对照品储备液1 ml置于50 ml容量瓶中,加乙醇稀释至刻度,摇匀,作为对照品溶液(每1 ml含亮蓝4.128 μg)。
2.4 供试品溶液的制备
精密称取上述3号染色的青黛药材粉末0.5 g,加甲醇25 ml,超声处理10 min,静置,放凉,用0.22 μm的微孔滤膜滤过,取续滤液作为供试品溶液。
2.5 线性关系考察
取“2.3”项下的对照品储备溶液作为对照品5号溶液,然后精密吸取5号对照品储备溶液3 ml置于10 ml容量瓶中,加乙醇稀释至刻度,作为4号对照品溶液;精密吸取4号对照品溶液3 ml置于10 ml容量瓶中,加乙醇稀释至刻度,作为3号对照品溶液;精密吸取3号对照品溶液3 ml置于10 ml容量瓶中,加乙醇稀释至刻度,作为2号对照品溶液;精密吸取2号对照品储备溶液3 ml置于10 ml容量瓶中,加乙醇稀释至刻度,作为1号对照品溶液。精密吸取上述1—5号对照品溶液各1 μl,按照“2.2”项下的色谱质谱条件进行测定,以进样量的质量为横坐标(X,ng),相对应的的色谱峰面积为纵坐标(Y)进行线性回归,结果亮蓝对照品的质量在16.27~2 008.27 ng范围内线性方程为Y=4.590 1X-25.823,相关系数r=1.000 0。
2.6 精密度试验
取“2.3”项下的1、3和5号对照品溶液,按照“2.2”项下的色谱条件分别进样1 μl,重复进样6次,记录峰面积。结果显示,亮蓝峰面积的RSD为0.38%,表明该仪器的精密度良好。
2.7 稳定性试验
取3号染色青黛药材,按照“2.4”项下供试品溶液制备,按照“2.2”项下的色谱条件,分别在0、1、2、4、8、12及24 h进行测定,进样量为1 μl,连续考察24 h,记录色谱峰面积。结果显示,亮蓝峰面积的RSD为2.06%,表明该供试品溶液在24 h内稳定性良好。
2.8 重复性试验
取3号染色青黛药材,按照“2.4”项下的供试品制备方法,平行制备6份供试品溶液,按照“2.2”项下的色谱条件进行测定。结果显示,6份供试品中亮蓝含量值的RSD为1.53%,表明该方法的重复性良好。
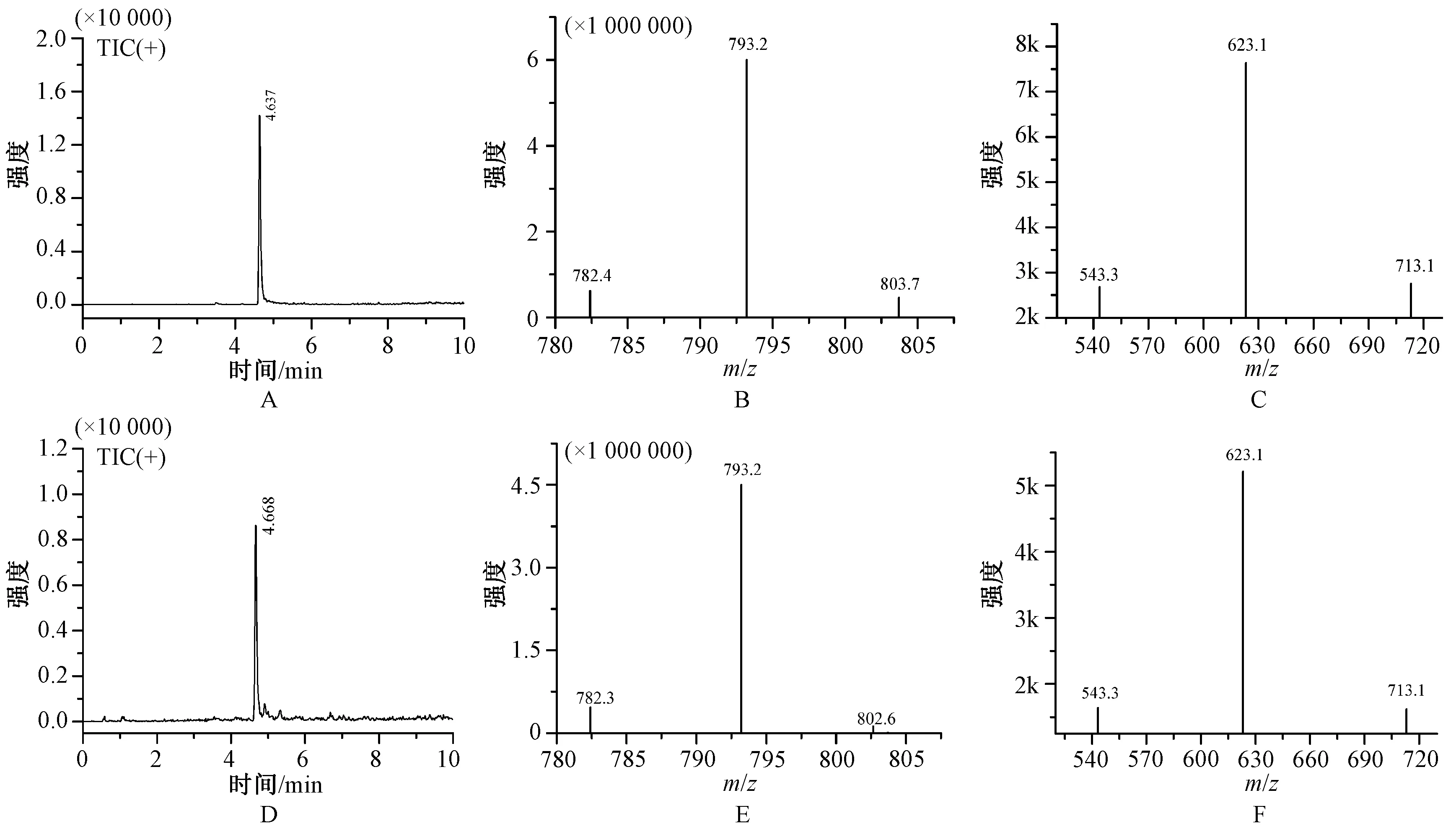
A.亮蓝对照品的总离子流图;B.亮蓝对照品的一级质谱图;C.亮蓝对照品的二级碎片质谱图;D.青黛样品中亮蓝的总离子流图;E.青黛样品中亮蓝的一级质谱图;F.青黛样品中亮蓝的二级碎片质谱图A. total ion current chromatogram of reference indigo naturalis; B. primary mass spectra of reference indigo naturalis; C. secondary fragment mass spectra of reference indigo naturalis; D. total ion current chromatogram of brilliant blue in indigo naturalis sample; E. primary mass spectra of brilliant blue in indigo naturalis sample; F. secondary fragment mass spectra of brilliant blue in indigo naturalis sample图2 青黛中亮蓝的质谱图Fig 2 Mass spectra chromatogram of brilliant blue in indigo naturali
2.9 加标回收试验
精密称取3号染色青黛药材0.25 g,共9份,每3份为1组,取“2.3”项下亮蓝对照品溶液1 ml置于25 ml容量瓶中,加乙醇稀释至刻度,作为待加溶液,分别采用低、中及高三种不同浓度进行加标回收实验,然后分别向上述每组样品中,精密加入上述待加溶液1、2及3 ml,然后按照“2.4”项下供试品溶液的制备方法制备和“2.2”项下色谱质谱条件进行测定,计算亮蓝的加标回收率,结果见表1。

表1 亮蓝的加标回收试验结果(n=9)Tab 1 Results of standard addition recovery of brilliant blue (n=9)
2.10 亮蓝的含量测定
取上述3号和8号两个不同产地的染色青黛药材,按照“2.4”项下供试品溶液的方法制备,按照“2.2”项下的色谱条件进行测定,用标曲法分别计算两批染色青黛药材中亮蓝的含量。结果显示,3号和8号样品中亮蓝的含量分别为1.230 6和2.012 8 mg/kg。
3 讨论
3.1 非法染色的鉴别
本研究首先将10批青黛药材进行提取处理,采用常用的青黛染色剂亮蓝、亮蓝G、酸性绿50、亚甲基蓝和甲基蓝等5种对照品,按照《中华人民共和国药典:四部》(2015年版)色素指导原则进行TLC鉴别,结果发现有2批样品在与亮蓝对照品TLC相同的位置上具有相同的颜色的斑点,因此判断这2批药材可能进行了亮蓝染色;然后,采用UFLC-MS/MS对其进行鉴别和确认,结果发现供试品在与亮蓝对照品的TIC图在相同的保留时间出现相同的色谱峰,且一级质谱图显示亮蓝的母离子m/z为793.2 [M+H]峰,二级质谱图的碎片离子分别m/z 为713.1、623.1和543.3等3个碎片离子,且3号和8号2批样品的质谱一级和质谱二级碎片图与亮蓝标准品一致,因此更加确定上述2批青黛药材进行了亮蓝的染色。
3.2 质谱条件的优化
本研究首先采用正负离子扫描亮蓝标准品,结果发现正离子下质谱响应比负离子下高2个数量级,母离子为 [M+H]峰为793.2,选择强度最高的二级特征离子[M-SO3]+(m/z 713.1)峰面积进行定量,采用MRM模式,进行准确定量,同时分别采用采用乙腈-水、乙腈-0.1%甲酸水溶液及甲醇-0.1%甲酸水溶液等流动相进行检测,结果发现甲醇-0.1%水溶液出峰时间太长,峰形偏宽;乙腈-水溶液色谱峰有点拖尾;乙腈-0.1%甲酸水溶液色谱峰形对称,基线平稳。故本研究最终确定乙腈-0.1%甲酸水溶液为流动相,对亮蓝进行了检测。
3.3 结果分析
从本研究所建立的方法来看,10批不同产地的青黛药材中就有2批药材进行了亮蓝染色,因此对青黛药材中的非法染色进行研究是有必要的。目前,青黛药材的国家药品补充检验批件中有非法染色孔雀石绿的检查项,未有亮蓝的染色检查项目。参考食品添加剂中对亮蓝的限量标准,最高标准为冰淇淋中的使用量不得超过0.022 g/kg,判断上述2批青黛中虽有添加亮蓝,但未超标。但由于药品中不允许检出亮蓝,故应引起检验监管部门的注意,同时建议将亮蓝的染色鉴别增加到青黛药材检验批件标准中。本研究所建立的亮蓝的UFLC-MSMS法通过方法学验证,具有灵敏度高、操作简单和准确度高的优点,可以为青黛药材中亮蓝的染色鉴别和含量测定提供参考。
