土壤环境对传统豆酱制酱过程中细菌群落演替的影响
2019-12-04安飞宇武俊瑞刘一鸣孙雪婷祝新媛魏丽丽乌日娜
安飞宇,武俊瑞,刘一鸣,孙雪婷,祝新媛,魏丽丽,乌日娜,*
(1.沈阳农业大学食品学院,辽宁 沈阳 110866;2.沈阳农业大学外语教学部,辽宁 沈阳 110866)
我国酱的起源非常早,最早甚至可以追溯到商周时期,而关于豆酱的起源,最初的记载始见于西汉[1]。传统自然发酵豆酱是以大豆为主要原料,经自然发酵而成的半流动状态的发酵食品[2]。自然发酵豆酱作为日常的调味品,因其独特的风味,深受广大消费者的喜爱。豆酱不仅具有丰富的营养价值,还具有抗氧化性、抑制血清胆固醇上升、抗癌、抗诱变性、降血压等保健功能[3-5],因此近年来自然发酵豆酱引起了学者们的广泛关注。
目前,工业上大多通过添加菌种来制作豆酱。但是菌种的单一性使得工业豆酱的风味及适口性远不如自然发酵豆酱[6]。传统发酵豆酱所具有的独特色、香、味、体是在复杂的微生物的共同作用下,通过许多的生化反应形成的,其中有蛋白质的水解、乙醇的发酵、淀粉的糖化、有机酸的形成、脂肪的水解等[7]。民间素来就有“百家酱来百家味”的说法,这说明传统自然发酵豆酱的风味多变。其原因是由于豆酱的生产原料、生产工艺及环境等因素不同,造成了自然发酵豆酱的菌群结构具有多样性,从而导致其风味的不同。
传统自然发酵豆酱的制作主要分为两个阶段,酱醅阶段和制酱阶段[8]。与酱醅阶段不同,在制酱阶段的发酵过程处于室外开放式环境中。由于传统农家自然发酵豆酱的酱缸都要放置在阳光充足、能保证其发酵温度的田地上,因此推断酱缸周围的土壤环境可能会对发酵豆酱的菌群结构产生一定影响。近年来,虽然国内外对自然发酵豆酱的群落组成做了大量的研究工作[2,9-11],但鲜见关于土壤环境对自然发酵豆酱微生物菌群结构的影响。目前,有关于土壤环境对发酵食品菌群结构影响的报道大多都集中于白酒窖泥及酒厂土壤的研究,已有报道称土壤环境会对白酒发酵过程中的微生物的数量和优势菌群产生影响,从而影响酒质的优劣[12-13]。因此,探究土壤环境对自然发酵豆酱细菌菌群结构影响具有重要意义。
随着高通量测序技术的不断进步,越来越多的科研人员利用该技术对复杂环境微生物进行分析。孙卫宁等[14]利用高通量测序技术对不同季节酿造的浓香型白酒的细菌多样性进行研究。沈馨等[15]利用Illumina MiSeq高通量测序系统对辣椒酱中核心菌群的群落特征进行了研究,为辣椒酱中细菌的研究提供了更准确、更科学的数据资源。本实验采用高通量测序技术分析了在发酵过程中传统自然发酵豆酱细菌菌群结构的变化,及其周围土壤环境和酱醅的细菌群落组成。揭示土壤环境对自然发酵豆酱细菌菌群结构影响,为提高豆酱风味及品质研究提供一定的理论参考。
1 材料与方法
1.1 材料与试剂
实验样品:采集自两家东北地区农家(辽宁四平S家、辽中L家)采用东北传统方法制作的成熟期酱醅(编号为SK、LK),发酵酱缸周围土壤样品(编号为ST、LT),以及各不同发酵阶段的豆酱样品各6 份,分别在发酵0、7、14、21、28、35 d(成熟)进行取样(分别编号为S1~S6、L1~L6),样品采集方式为3 个不同位点混合采样。样品采集后,放置于冰盒中,并迅速转移至-80 ℃冰箱低温冻藏。
E.Z.N.A.® Soil试剂盒 美国Omega公司;聚合酶链式反应(polymerase chain reaction,PCR)引物 上海美吉公司;AxyPrep DNA凝胶提取试剂盒 美国Axygen公司。
1.2 仪器与设备
ABI GeneAmp® 9700型PCR仪 美国ABI公司;QuantiFluorTM-ST 美国Promega公司;Illumina MiSeq PE300测序平台 美国Illumina公司。
1.3 方法
1.3.1 样品总DNA的提取
根据Sun等[11]的方法提取各样品的总DNA,利用NanoDrop 2000对DNA浓度和纯度进行检测。1%琼脂凝胶电泳检测DNA的完整性。将浓度和纯度均合格的DNA在-20 ℃条件下保存,用于PCR扩增。
1.3.2 PCR扩增
用338F(5’-ACTCCTACGGGAGGCAGCAG-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)引物对V3-V4可变区进行扩增,扩增程序:在95 ℃预变性3 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,27 个循环;最后72 ℃延伸10 min。
1.3.3 Illumina MiSeq测序及数据处理
使用2%琼脂糖凝胶回收PCR产物,利用凝胶提取试剂盒进行纯化,Tris-HCl洗脱,2%琼脂糖凝胶电泳检测。利用QuantiFluor™-ST进行检测定量。根据Illumina MiSeq平台标准操作规程将纯化后的扩增片段构建PE 2*300文库[16]。最后利用Illumina公司的MiSeq PE300平台进行测序[17]。运用MiSeq工具中的MiSeq Control Software(MCS)进行碱基识别和图像分析,数据下机后在Illumina basespace云端计算平台进行初始分类分析。基于分类学信息,在各分类学水平上进行群落结构等深入的统计学和可视化分析。
2 结果与分析
2.1 稀释曲线
稀释曲线主要利用各样本的测序量在不同测序深度时的微生物多样性指数构建曲线,可用来说明样本的测序数据量是否合理。如图1所示,当测序深度<10 000时,多样性指数随着测序深度的增加而迅速增加;当测序深度在10 000~30 000之间时,多样性指数随着测序深度的增加而缓慢增加;测序深度>30 000时,稀释度曲线趋于平台期,表明测序准确有效,可以充分反映样品的多样性。

图1 样品稀释曲线Fig. 1 Rarefaction curves for samples
2.2 样品复杂度分析
样品的复杂度即α多样性反映微生物的多样性和群落的物种丰富度,包括OTUs数、Shannon指数、ACE指数、Chao1指数等结果[8]。ACE指数和Chao1指数都是用来计算菌群丰度,ACE指数或Chao1指数越大,表明群落的丰富度越高。如表1、2所示,与酱醅和豆酱样品相比,土壤样品的物种最为丰富,菌群结构明显复杂。两家酱醅的群落丰富度比较相似,进入液态酱阶段后,物种丰度都有所上升,随着发酵的进行有所下降,随后丰度上下波动。特别是,两家豆酱的细菌丰度指数都在第4个发酵阶段(S4、L4)出现峰值,说明该阶段可能为传统自然发酵豆酱发酵最为活跃的时期。
通过Shannon指数对样品多样性的比较,发现两家酱醅中菌群的多样性指数均低于下酱当天的样品,可能的原因为:在下酱的过程中,酱缸周围土壤环境中的微生物扩散进入酱缸,从而影响了豆酱发酵初期的细菌多样性。同时,与下酱当天的样品相比,各发酵阶段豆酱的菌群多样性整体呈下降趋势。此前,虽有学者研究表明窖泥和糟醅具有较高相似度的细菌区系,其间存在较大规模的细菌相互扩散[18-19],但鲜有有关环境土壤对发酵豆酱菌群结构影响的报道。另外,所有土壤,酱醅及豆酱样品的Coverage值都接近于1,说明覆盖率较高,测序结果体现了样本中微生物的真实情况。
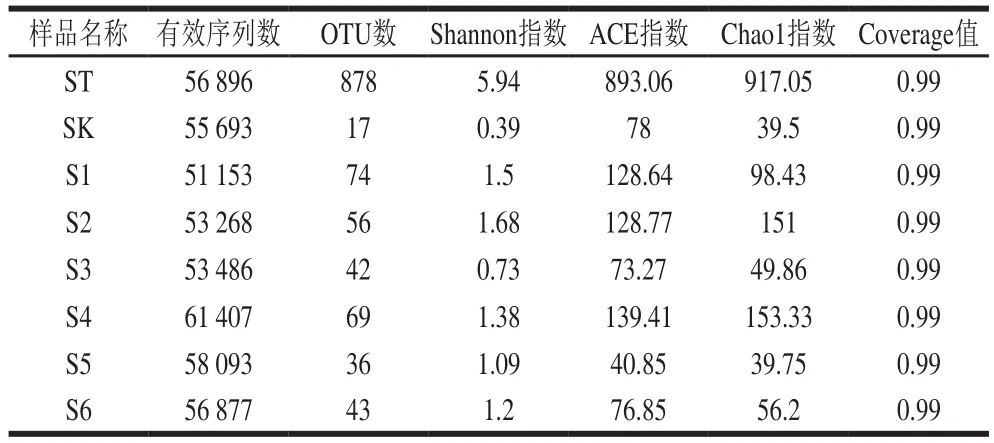
表1 四平样品α多样性分析Table 1 α Diversity in samples collected from Siping

表2 辽中样品α多样性分析Table 2 α Diversity in samples collected from Liaozhong
2.3 物种组成比较分析
2.3.1 物种Venn图分析
Venn图可用于统计多个样本中所共有和独有的物种(如OTU)数目,可以比较直观地表现样本的物种组成相似性及重叠情况[20]。图2A为四平发酵前3 个阶段的豆酱与酱醅及土壤的Venn图,样品S1、S2、S3与土壤单独共有(排除与酱醅共有)的OTU数分别为27(36.49%)、15(26.79%)、7(16.67%)。到了发酵后3 个阶段,如图2B所示,单独共有OTU数减少为24(34.78%)、7(19.44%)、10(23.26%)。图3A为辽中豆酱发酵前3 个阶段与酱醅及土壤的Venn图,样品L1、L2、L3与土壤单独共有的OTU数分别为47(40.51%)、9(21.95%)、13(25%)。到了发酵后3 个阶段,如图3B所示,单独共有的OTU数减少为11(23.40%)、5(11.90%)、18(28.13%),变化趋势与四平样品结果相似。
结果表明,两家豆酱在发酵的第1个阶段(S1、T1),豆酱与土壤单独共有OTU数目及占比均达到峰值,说明此阶段土壤环境对发酵豆酱的OTU影响最大。随着发酵的进行,这种影响逐渐减弱并上下波动,且与多样性指数分析结果一致。可能的原因为:在发酵的第1个阶段,也就是下酱时期,土壤环境与豆酱直接接触的机会最多,存在较大规模的细菌相互扩散,从而对豆酱初期的菌群结构产生了一定影响。后期随着发酵的进行,菌群结构会趋于相对稳定[6,21],同时土壤环境与豆酱接触的机会减少,因此土壤环境对后期豆酱菌群结构影响较小。

图2 四平样品Venn图Fig. 2 Venn diagrams for Siping samples
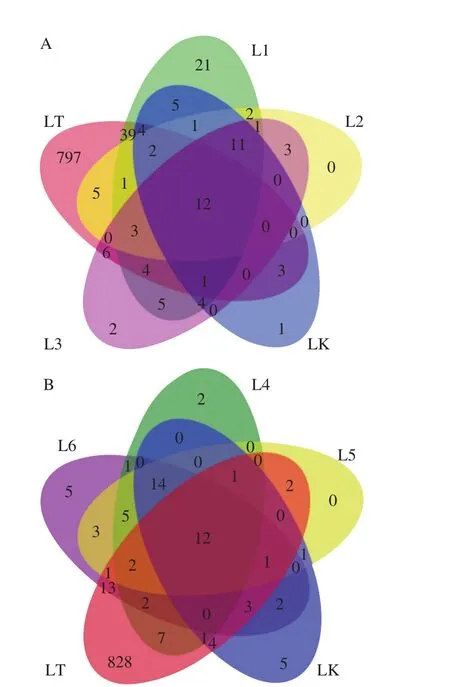
图3 辽中样品Venn图Fig. 3 Venn diagrams for Liaozhong samples
2.3.2 门水平下样品菌群结构分析
根据分类学分析结果,可以得知不同样本在各分类水平上的群落结构组成情况。四平样品在门水平上的群落组分如图4所示,土壤中微生物菌群结构最为复杂,其主要的细菌门包括:厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)、酸杆菌门(Acidobacteria)、芽单胞菌门(Gemmatimonadetes)。发酵酱醅和豆酱的菌群结构在门水平上较为相似,其主要的细菌门包括:厚壁菌门、变形菌门、放线菌门。发酵不同阶段的优势细菌门均为厚壁菌门(占总数86.72%~99.50%),同时在酱醅阶段及发酵前期,变形菌门(占总数10.84%~12.87%)也被大量检出。值得一提的是,在豆酱发酵的各个阶段均检测到一定数量的放线菌门(0.35%~12.87%),并在发酵前两个阶段最为丰富,且放线菌门并未在酱醅样品中大量发现。
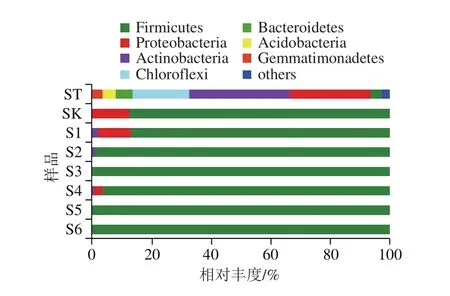
图4 门水平下四平样品群落组分图Fig. 4 Microbial community composition at the phylum level in Siping samples
辽中样品也出现了相似结果,如图5所示,辽中样品主要的细菌门包括:厚壁菌门、变形菌门、放线菌门、绿弯菌门、酸杆菌门、芽单胞菌门、拟杆菌门,其中放线菌门的相对丰度低于四平样品。发酵酱醅和豆酱的菌群结构在门水平上也较为相似,其主要的细菌门包括:厚壁菌门、变形菌门、放线菌门。发酵不同阶段的优势细菌门均为厚壁菌门(占总数84.46%~99.86%),同时在发酵的首个阶段,变形菌门(14.10%)和放线菌门(1.40%)也被大量检出,但在酱醅样品中放线菌门并未大量出现。因此,推测土壤环境的确会对豆酱菌群结构产生一定影响,且主要的扩散菌门为放线菌门。但随着发酵的进行,这种影响会逐渐减弱,并维持在一定水平,该结果与之前的Venn图分析结果一致。另外由图4、5可知,辽中土壤对其豆酱的菌群结构影响较小,可能是由于辽中土壤样品中的放线菌门的相对丰度(23.90%)要低于四平样品(33.91%)导致。

图5 门水平下辽中样品群落组分图Fig. 5 Microbial community composition at the phylum level in Liaozhong samples
2.3.3 属水平下样品菌群结构分析
进一步分析了属水平下各样品的菌群结构,动态跟踪了整个豆酱发酵过程中细菌变化过程。如图6所示,四平样品主要的细菌菌属为:芽孢杆菌属(Bacillus)、四联球菌属(Tetragenococcus)、明串珠菌属(Leuconostoc)、假单胞菌属(Pseudomonas)、乳杆菌属(Lactobaillus)、魏斯氏菌属(Weissella)、不动杆菌属(Acinetobacter)、考克氏菌属(Kocuria)等。
在酱醅阶段,优势菌属为:芽孢杆菌属(87.08%)和假单胞菌属(12.86%)。加入盐水后,进入豆酱阶段,其优势菌属为:芽孢杆菌属(12.56%~68.62%)、四联球菌属(0.0 4%~8 1.4 3%)、明串珠菌属(0.89%~10.40%)、魏斯氏菌属(0.09%~7.26%)、乳杆菌属(0.65%~5.94%)。酱块加入盐水后,前3 个阶段芽孢杆菌逐渐减少,由S1阶段的68.62%减少到S3的12.56%。在豆酱中,具有代表性的芽孢杆菌为枯草芽孢杆菌(Bacillus subtilis)和地衣芽孢杆菌(Bacillus licheniformis)[22],减少的原因可能是随着发酵的进行,发酵体系内的乳酸菌产乳酸使总酸升高,pH值下降,由于较低的pH值对地衣芽孢杆菌的生长产生抑制作用[23],所以芽孢杆菌的含量逐渐减少。原本在酱醅中大量存在的假单胞菌属在豆酱发酵阶段逐渐减少,在S3阶段仅占0.06%,假单胞菌属为导致食品腐败的重要腐败细菌,由于其为严格需氧型细菌[24],豆酱发酵过程中的无氧环境不利于假单胞菌属的生长。四联球菌在S2阶段迅速增加,到S3阶段成为豆酱中占绝对优势的细菌。四联球菌属是促进健康的益生菌,它广泛存在于豆酱、酱油、鱼酱等发酵食品中。酵母菌参加代谢反应的产物主要为醇类和醛类,而四联球菌属代谢的产物则主要是以乳酸为主要酸的多种有机酸类,两者结合,可以显著提高产品中酯类物质含量,所以四联球菌属在改善豆酱风味方面具有重要作用[25]。
相较于发酵豆酱而言,土壤中的细菌菌属过于复杂,单从群落组分图上无法准确得知其对豆酱菌群的具体影响。因此运用Heatmap图进行深入分析,Heatmap图是以颜色梯度表征二维矩阵或表格中的数据大小,并呈现群落物种组成信息。可使高丰度和低丰度的物种分块聚集,通过颜色变化与相似程度反映不同样本在各分类水平上群落组成的相似性和差异性[26],如图7所示,放线菌门中的考克氏菌属[27](0.28%~1.57%)仅在土壤及豆酱样品中(尤其是发酵前期)有较高丰度,而在酱醅样品中并未大量发现。有报道称,考克氏菌属是自然发酵腐乳中的优势菌属之一,其菌株胞外蛋白酶可以水解大豆蛋白产生氨基酸等风味物质[28-29]。同时这也再次印证了,土壤环境的确会对豆酱菌群结构产生一定影响,且主要的扩散菌门为放菌门的这一观点。

图6 属水平下四平样品群落组分图Fig. 6 Microbial community composition at the genus level in Siping samples
如图8所示,辽中样品主要的细菌菌属为四联球菌属、乳杆菌属、明串珠菌属、肠球菌属(Enterococcus)、假单胞菌属、不动杆菌属、魏斯氏菌属等。在酱醅阶段,优势菌属为乳杆菌属(88.16%),明串珠菌属(8.59%)。加入盐水后,进入豆酱阶段,其优势菌属为四联球菌属(0.29%~96.58%)、乳杆菌属(1.96%~51.85%)、肠球菌属(0.26%~12.63%)、明串珠菌属(0.5 5%~1 1.0 6%)、假单胞菌属(0.04%~7.90%)。两家豆酱中都存在乳杆菌属和明串珠菌属,豆酱中的乳杆菌主要是植物乳杆菌[30],其不仅有利于食品的发酵,还可以改善食物的风味,还具有一定益生特性,包括维持肠道内菌群平衡、抑制肿瘤细胞的形成、降低血清胆固醇等[31-32]。明串珠菌是革兰氏阳性、耐氧的一种乳酸菌,肠膜明串珠菌产乳酸,可以在高盐和高糖环境中生长,因此在豆酱中广泛存在[33]。此外,肠球菌属仅为辽中豆酱的优势菌属,而在四平豆酱中并未检出,其可能的原因为:Jeong等[34]发现在酱块发酵过程中,随着芽孢杆菌数量增多肠球菌数量降低,两种菌种可能存在负相关关系,而四平豆酱中芽孢杆菌为优势细菌,因此导致其肠球菌属丰度降低。

图7 属水平下四平样品Heatmap图Fig. 7 Heatmap diagram at the genus level for Siping samples
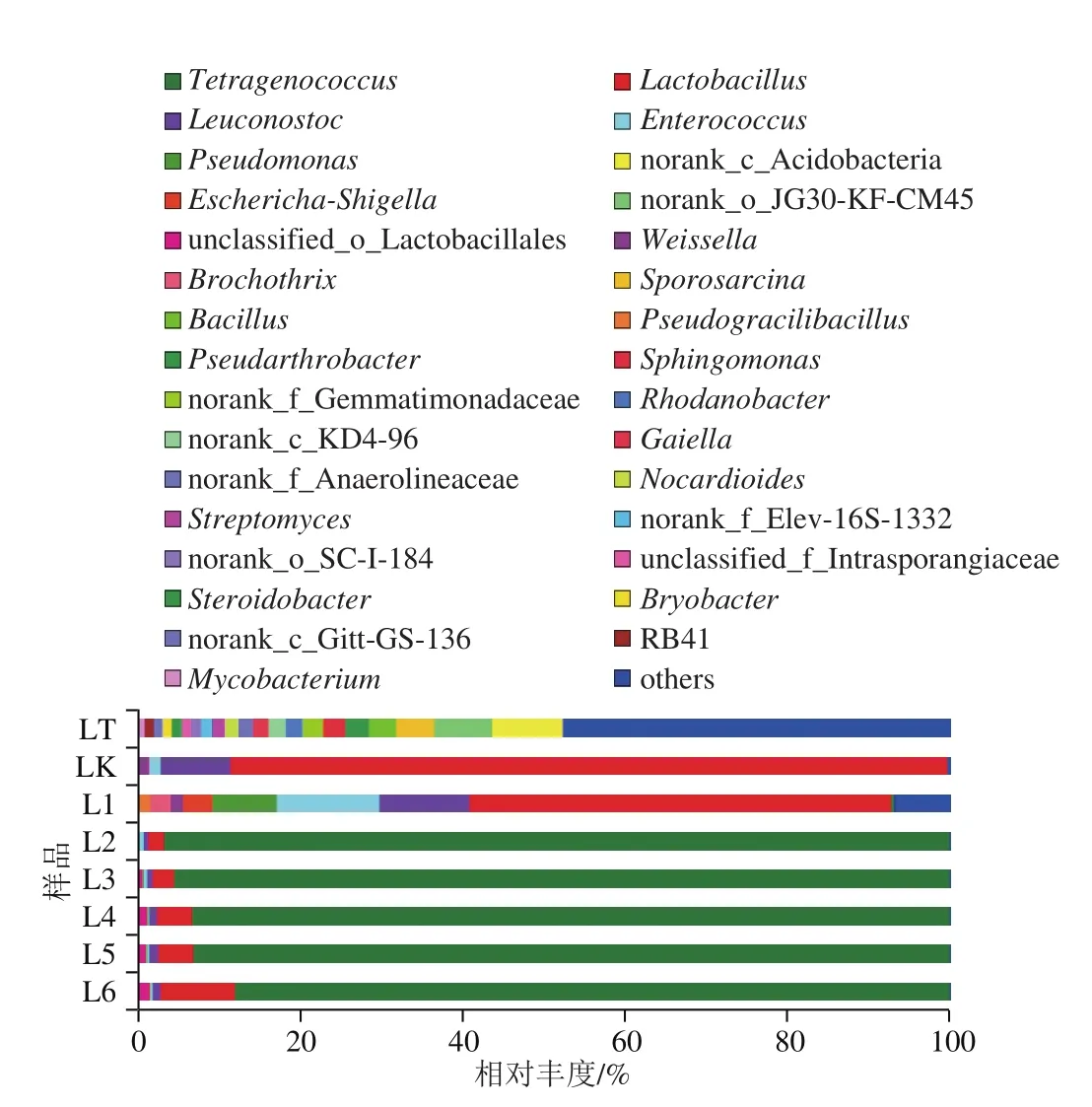
图8 属水平下辽中样品群落组分图Fig. 8 Microbial community composition at the genus level in Liaozhong samples
图9 为辽中样品Heatmap图,在丰度前20的菌属中并未找到土壤与豆酱有明显相关性的菌属,其原因可能为:1)下酱过程中操作手法的不同,使土壤环境与豆酱接触少;2)辽中土壤样品中的放线菌门的相对丰度较低,无法深入影响发酵豆酱菌群结构或是所影响的菌属丰度较低,无法在图中体现。该结果也与之前门水平的研究结果一致。两家豆酱细菌群落结构有较大差异,表明传统发酵豆酱中细菌菌群结构组成与原料、制作手法、地区差异均有密切联系,因此还需要进行后续实验和数据积累,并结合如转录组、代谢组等其他组学共同研究,以期进一步了解传统自然发酵豆酱风味和微生物组成差异的原因。
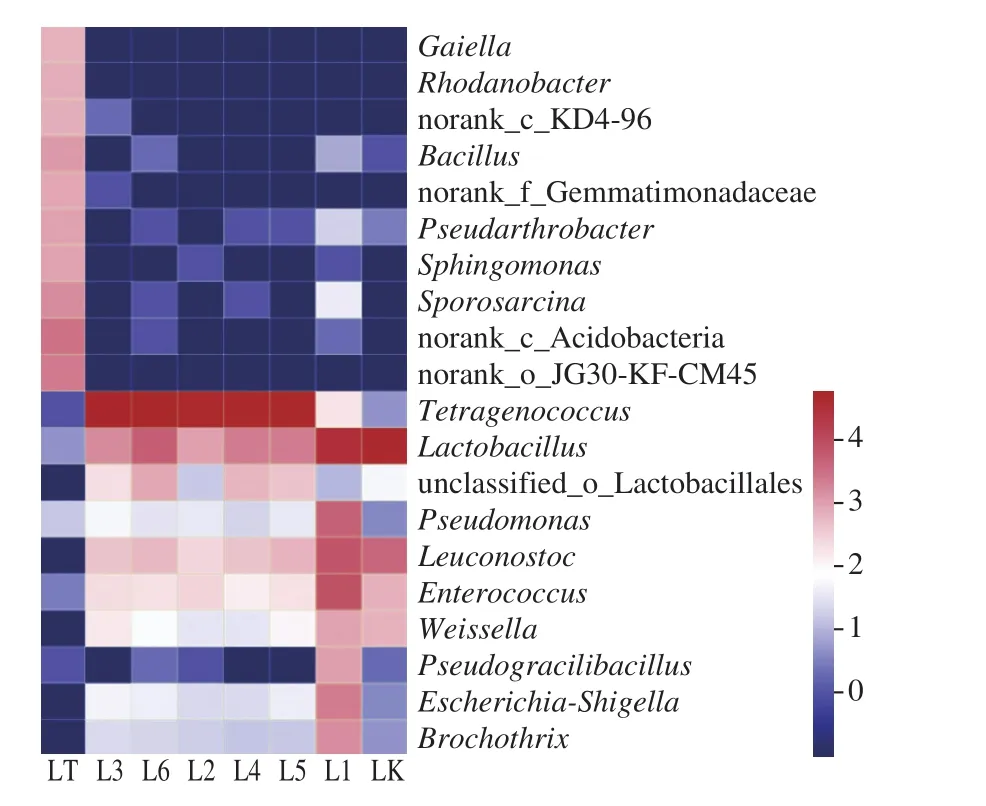
图9 属水平下辽中样品Heatmap图Fig. 9 Heatmap diagram at the genus level for Liaozhong samples
3 结 论
利用高通量测序技术,分别鉴定了采集自四平,辽中两家传统自然发酵的成熟酱醅、豆酱以及酱缸周围土壤环境的细菌群落结构。结果表明,四平样品酱醅阶段优势菌属及相对丰度为芽孢杆菌属(87.08%)和假单胞菌属(12.86%)。豆酱阶段优势菌属及相对丰度为芽孢杆菌属(12.56%~68.62%)、四联球菌属(0.0 4%~8 1.4 3%)、明串珠菌属(0.89%~10.40%)、魏斯氏菌属(0.09%~7.26%)、乳杆菌属(0.65%~5.94%)。辽中样品酱醅阶段优势菌属及相对丰度为乳杆菌属(88.16%),明串珠菌属(8.59%)。豆酱阶段优势菌属及相对丰度为四联球菌属(0.29%~96.58%)、乳杆菌属(1.96%~51.85%)、肠球菌属(0.2 6%~1 2.6 3%)、明串珠菌属(0.55%~11.06%)、假单胞菌属(0.04%~7.90%)。在揭示自然发酵豆酱细菌菌群的动态变化的同时,比较分析土壤环境对自然发酵豆酱细菌菌群结构的影响,本研究发现:土壤环境会对豆酱菌群结构产生一定影响,且主要的扩散细菌为放线菌门中的考克氏菌属,但随着发酵的进行,这种影响会逐渐减弱,并维持在一定水平。
4 讨 论
高通量测序结果表明,两家豆酱细菌群落结构存在一定差异,其可能是除原料及酿造工艺外造成豆酱风味不同的另一主要原因。土壤环境对豆酱细菌菌群结构产生一定影响,但随着发酵的进行,豆酱菌群结构会趋于相对稳定,土壤环境与豆酱接触的机会减少,从而使得土壤环境对豆酱细菌菌群结构影响减小。同时,下酱过程中的操作手法,土壤样品中的放线菌门的相对丰度也可能对菌群结构变化造成影响。此外,土壤环境中的霉菌对豆酱菌群结构是否存在影响还有待探讨,同时还需结合如转录组、代谢组等其他组学共同研究,以期进一步了解传统自然发酵豆酱风味和微生物组成差异的原因及其代谢机理。
