Colon cancer stemness as a reversible epigenetic state: Implications for anticancer therapies
2019-11-28AudreyVincentchaOuelkditeOumouchalMouloudSouidiJulieLeclercBernadetteNeveIsabelleVanSeuningen
Audrey Vincent,Aïcha Ouelkdite-Oumouchal,Mouloud Souidi,Julie Leclerc,Bernadette Neve,Isabelle Van Seuningen
Audrey Vincent,Aïcha Ouelkdite-Oumouchal,Mouloud Souidi,Julie Leclerc,Bernadette Neve,Isabelle Van Seuningen,Lille University,Institut National de la Santé et de la Recherche Médicale,CHU Lille,UMR-S 1172-Jean-Pierre Aubert Research Center,Lille F-59000,France
Julie Leclerc,Department of Biochemistry and Molecular Biology,Lille University Hospital,Lille F-59000,France
Abstract
Key words: Cancer stem cells; Colon cancer; Epigenetics; Chromatin modifying enzymes;CD44; CD133; CD166
INTRODUCTION
Hierarchy of the tumor: turning an old concept into a new dogma
Although only recently upgraded as the keystone of the natural history of tumors,the concept of “cancer stem cells (CSCs)”was anticipated several decades ago as researchers soon discovered that cancer cells possessed unequal capacities when it comes to initiating a new tumor or resisting to therapies[1].Indeed already during the 1960s,ethically disputed experiments of auto-transplantation that were conducted in human patients demonstrated that numerous cancer cells were necessary to establish cancer transplants,giving hints on the rare nature (1/1000000) of tumor-initiating cells[2].
With the arrival of the first commercially available cell sorters,followed by immunocompromised mouse models that allowed selective xenotransplantation of cancer cells,the interest in this cancer cell subpopulation has then been growing exponentially,with the field of hematologic malignancies as pioneers[1-3].As early stem or progenitor cells were shown to be involved in leukemias and myeloproliferative disorders,tumor initiating cells have rapidly been renamed “cancer stem cells”,hence creating a link with histological observations from the 1850’s when pathologists had first hypothesized that tumors could develop from residual embryonic tissues[1-3].Indeed,CSCs share numerous characteristics with normal embryonic stem cells,such as rareness,cell cycle arrest and quiescence,unlimited selfrenewal through asymmetric division,and addiction to stem cell signaling pathways.
In solid tumors,the cancer stem cell (CSC) model (Figure 1B) was initially considered as a concept that could not be applied to all tumor types and was often opposed to the stochastic clonal evolution hypothesis[4,5],where genetic mutations are the major cause of tumor heterogeneity (Figure 1A)[6,7].Increasing evidence of cancer plasticity,where cells easily exchange their position in the tumor hierarchy,switching from stem to non-stem states[8,9]and also from non-stem to stem states,reconcile these two models (Figure 1C).Indeed,several studies have demonstrated that cancer cells from different types of tumors,including colon cancer,can naturally convert to CSCs in culture,in total absence of therapeutic agents inducing genetic alteration[8].Additionally,anti-cancer treatments such as chemotherapies[10]or radiotherapy[9]not only participate in the selection of resistant clones in the bulk of a tumor but also induce stemness characteristics in non-stem cancer cells.These findings are transposable to tumors from patients in whom stemness-related aggressiveness(invasion capacities,release of circulating tumor cells) is either innate or acquired after exposure to hypoxia,metabolic stress,and treatments.
More importantly,the extreme cellular plasticity involving rapid phenotype switches between CSCs and their non-stem counterpart is probably mediated by epigenetic mechanisms that are reversible in nature,rather than newly acquired genetic mutations.Indeed,we (unpublished data) and others have shown a systematic equilibrium between CSC marker expressing and non-expressing cells that spontaneously occurs after cell sorting of negativevspositive populations[11].In accordance with epigenetic mechanisms involved in this balance between stem and non-stem cancer cells,CSCs harbor a permissive epigenetic state[12-14],comparable to normal stem cells,while epigenetic profiles of differentiated cells are locked in order to shape cellular identity and functions.However,numerous genetic alterations may render cancer cell reprogramming more complicated to target.Understanding this flexibility is crucial for the development of new anticancer drugs.Therefore,new therapeutic strategies will have to combine the targeting of the bulk of the tumor and of the CSCs,whether they are pre-existing or induced.Hence,if these different types of CSCs share the same reversible reprogramming mechanisms,epigenetic therapies would represent an interesting strategy (Figure 2).
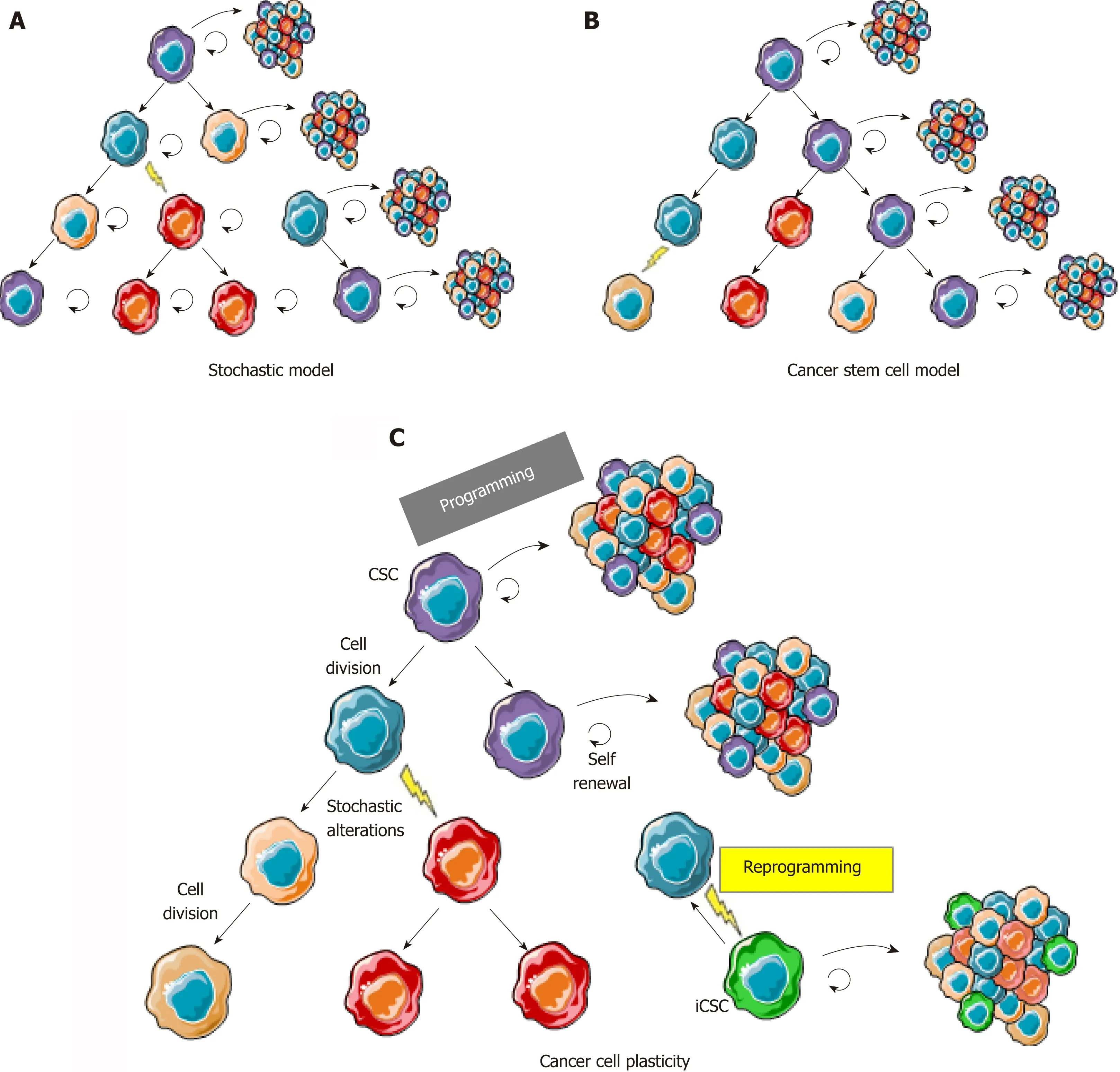
Figure 1 The cancer cell plasticity model reconciles cancer stem cell and stochastic models.
UNRAVELING THE EPIGENETIC SIGNATURE OF CSCs: A KEY TO UNDERSTANDING CANCER CELL PLASTICITY AND REPROGRAMMING
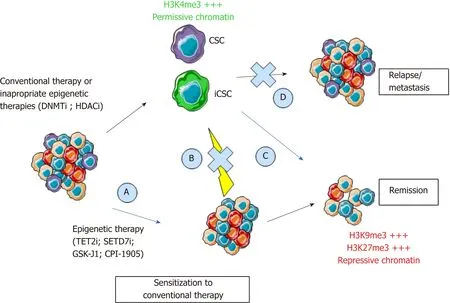
Figure 2 Epigenetic programming and reprogramming of cancer cells and consequences for therapeutic strategies.
Current research on induced pluripotent stem cells teaches us that erasing epigenetic marks of the differentiated cell of origin greatly improves reprogramming[15,16].Mapping stemness-associated chromatin modifications would surely facilitate the development of therapeutic strategies evoking differentiation of CSCs.Indeed,the“differentiating strategy” has proven its efficiency in certain types of hematologic tumors years ago[17].On the other hand,these strategies have failed to prove their systematic efficacy in solid tumors,where CSCs may come from multiple origins,including normal differentiated cells[8,18],or stochastic genetic events altering cancer cells along tumor evolution.
Molecular mechanisms involved in the shaping of the cancer epigenetic landscapes,and especially in CSCs,are complex.Genetic alterations leading to loss or gain of epienzyme functions have been described[19],but only rare studies focus exclusively on CSCs.Furthermore,overexpression of epienzymes may not reflect an oncogenic role.The histone methyltransferase enhancer of zeste 2 (EZH2) is the perfect example of this paradox,while its overactivation in certain types of cancers is the sole sign of a compensation mechanisms in cells where histone H3 K27 trimethylation is diluted over excessive proliferation[20-22].
Because of the rareness and diversity of CSCs and the fact that no consensus has been found for markers that would allow their proper isolation,few studies have been able to define clearly the cancer stemness-associated epigenetic profiles.It has been shown,however,that mammary and hepatic CSCs harbor more permissive chromatin profiles,more prone to gene activation,than non-stem cancer cells[12].They also harbor decreased DNA methylation and trimethylation of lysine 27 on histone H3 at tumor suppressor genes[12].Similarly,trimethylation of lysine 4 on histone H3 is found preferentially at pluripotency genes such as BMI1,NOTCH1,and WNT1 in CSCs from acute myeloid leukemia patients[13].CSCs from head and neck carcinomas harbor an epigenetic signature with only 22 differentially methylated genes between cluster of differentiation (CD)-44+ CSCs and CD44 non-stem cancer cell populations[14],pointing out subtle and specific differences between stem and non-stem cancer cells.The same type of signature has been identified in breast tumors[23],but still needs to be defined for CSCs from the different colon cancer molecular subtypes.
The common findings from studies on CSC epigenetic profiles are that CSC markers are either regulated by epigenetic mechanisms in normal and/or cancer cells or harbor different epigenetic profiles between stem and non-stem cancer cells[24].Alternatively,CSC markers can themselves be directly or indirectly responsible for chromatin modifications through their presence in Polycomb Repressive Complexes(BMI1) or through histone demethylation (JARID1B).
Among CSC markers,CD133 and CD44 have been extensively utilized to isolate cancer cells with tumorigenic characteristics in numerous types of cancers,including colon cancers in which CD133 predicts low survival.In combination with CD166,these two markers better stratify low,intermediate,and high-risk cases of colorectal cancer[25](CRC) than the three markers alone.We have shown that combined expression of these three markers is associated with stemness and resistance to 5-fluorouracil (5-FU) in colon cancer cells[26,27].Interestingly,expression and splicing of these three markers are epigenetically regulated in cancer cells.
Epigenetic regulation of PROM1,encoding the CSC marker CD133
CD133 is a 120 kDa transmembrane glycoprotein that was initially identified in hematopoietic stem cells[28]and is involved in cell-cell interactions and membrane organization,through its binding to phospholipids[29].CD133 is now used as a stem cell marker in most solid tumors including colorectal cancers[29].More importantly,CD133 is directly involved in stemness properties as its inhibition alters self-renewal and tumorigenic capacities[30].CD133 is also associated with metastasis and invasiveness through the decrease of metalloprotease 2 expression.Interestingly,its expression is positively correlated with the expression of ATP-binding cassette (ABC)transporters ABCG1 and ABCG2,hence associating CSC properties to chemoresistance through the presence of multidrug efflux pumps[28].CD133 is correlated to poor prognosis in numerous cancers including CRC.
The human PROM1 gene,which encodes CD133 (prominin-1),consists of 28 exons and is localized on chromosome 4p15.The regulation of PROM1 transcription includes five alternative promoters (P1-5) involved in embryonic phase development.PROM1 harbors seven alternative spliced variants,of which the most documented are CD133s1 and CD133s2 (lacking exon 3)[31,32].Of those only CD133s1 is mainly associated with normal tissue in brain,bone marrow,and blood[31].CD133s2 expression is widely observed in human fetal tissue and adult tissues and in several cancers,including breast,colon,lung,and pancreatic carcinomas.CD133s2 is also associated with the human stem cell niche[33].
PROM1 expression is inversely correlated with methylation of CpG islands in its promoter in numerous cancer cell lines[34,35].For example,in glioma tissues,an inverse correlation has been shown between the CpG methylation status of promoter P1 and P2 and expression levels of PROM1 transcripts.Epigenetic regulation of PROM1 also includes histone modifications,since synergistic effects are observed when using histone deacetylase (HDAC) inhibitors in combination with DNA methyltransferase(DNMT) inhibitors to re-express the cell surface marker CD133 in ovarian cancer cells[24].
Epigenetic regulation of CD44
CD44 is a transmembrane glycoprotein interacting with components of the extracellular matrix including hyaluronic acid,collagens,fibronectins,integrins,and laminin[36].These interactions induce cytoskeleton modifications and activation of signaling pathways involved in cell adhesion and migration.CD44 expression has been associated with tumor progression,epithelial-to-mesenchymal transition[37],and poor survival in colon cancers[38].Mutations have been described in solid tumors,suggesting its implication in carcinogenesis[37].Most importantly,CD44-variant-6 (v6)is a well-recognized marker of colon and gastric CSCs[39,40].
The human CD44 gene consists of 20 exons and is located on chromosome 11p13.Exons 1-5 and 15-19 encode homologous N-ter (extracellular) and C-ter (extracellular,transmembrane and intracellular) domains respectively forming the standard isoform CD44s.Alternative splicing of exons 5a-14 result in different variants/isoforms of CD44 (CD44v).CD44 variants are overexpressed in numerous types of solid tumors including pancreatic (CD44v2-6),breast (CD44v6/v8-10),prostate (CD44v2/v6),head and neck (CD44v3),and colon (CD44v6/v10) cancers[37].In contrast with CD44s variant that is absent from mouse normal intestinal stem cells[37],CD44 variants(CD44v4-10) have been associated with normal and cancer stemness.For instance,CD44v6 and CD44v4 are largely overexpressed in stem cells compared to their progeny (transit-amplifying cells).CD44 variants,and not CD44s,are involved in adenoma formation in mouse models of familial polycystic adenomas[41].Similarly,expression of CD44v6 is restricted to colon CSCs and is associated with worse survival in patients with CRC[39].In most studies,CD44v4-10 variants are associated with aggressiveness,resistance,metastasis,and poor prognosis in solid tumors including colon cancers.
Epigenetic regulation of the CD44 gene has recently been described.DNA methylation at CpG islands located in the promoter and histone H3 acetylation regulate its silencing or expression[37],respectively.DNMT inhibition induced DNA methylation and histone modification changes at the CD44 gene promoter,increasing CD44 mRNA levels in cancer cell lines[37,42].More importantly,alternative splicing of CD44 and,hence,the expression of CSC specific variants is epigenetically regulated.Indeed,accumulation of histone H3 lysine 9 trimethylation and HP1 stabilizes premRNA binding to the chromatin and therefore facilitates exon inclusion[43].
Epigenetic regulation of ALCAM encoding the CSC marker CD166
CD166 is a member of the immunoglobulin superfamily and is engaged in homophilic or heterophilic interactions with the cell surface receptor CD6.CD166,which is expressed on antigen-presenting cells,is involved in maturation of CD6-expressing resting T-cells and is also expressed in mesenchymal stem cells,neural cells,osteoblasts,and stromal cells of the bone marrow.It is involved in hematopoiesis,development of central and peripheral nervous system,sense organs,and differentiation of endothelial as well as epithelial lineages[44].CD166 has proven its relevance as a CSC marker alone or in combination with CD44 in several studies including studies on colon cancer cell lines[45,46].
The human gene ALCAM,encoding CD166,is located on chromosome 3q.13 and consists of 16 exons.A soluble isoform,produced through alternative splicing,has been described,but its role remains unknown[47].
The ALCAM promoter harbors several CpG islands regulated by DNA methylation.It has been shown that the DNMT inhibitor 5-Aza-2’-deoxycytidine increased its expression in breast cancer cells[48],hence raising questions about the use of these inhibitors in breast cancer patients.
Interestingly,the three discussed CSC and survival markers (CD44,CD166,and CD133,Figure 3) are not only epigenetically regulated in cancer cells,but our transcriptomic analyses of public CRC data also revealed that the combined expression of these markers in colon cancer is correlated with a specific panel of epienzyme expression (both positive and negative correlations are listed in Tables 1-6).
EPIENZYME CORRELATION WITH COLON CSC MARKERS:A HINT FOR SUCCESS IN EPIGENETIC THERAPEUTIC STRATEGIES?
Current epigenetic strategies
Most solid tumors,including CRC,acquire chemoresistance over time.In addition to expected chemo-induced genetic alterations,the molecular mechanisms involved include transcriptional plasticity that is regulated epigenetically,for example by multiple DNA methylation changes at CpG islands[49].Contrary to genetic alterations,epigenetic modifications are potentially reversible,paving the way for novel cancer therapies.
This past decade has seen the emergence of many epigenetic therapies,especially DNA hypomethylating drugs (DNA methyltransferase inhibitors) and HDAC inhibitors (HDACi),as well as lysine-specific histone demethylase-1,EZH2 inhibitors,and many others[50].
Epigenetic drugs have shown beneficial effects for the treatment of hematological malignancies and led to the approval of epidrugs like 5-azacitidine,decitabine,vorinostat,romidepsin,belinostat,and panobinostat for patient treatment[50].In contrast,clinical trials assessing the efficacy of these epigenetic drugs in monotherapies for CRC and other solid tumors failed to improve clinical outcomes with,in some cases,no response at all[51],never passing the phase III trial necessary for approval (clinical trials for CRC listed in Tables 1-6).
Several hypotheses could be raised regarding this apparent lack of efficacy of epidrugs for solid tumors.First,compared to hematologic malignancies,solid tumors harbor a weaker penetrance of mutations in genes encoding chromatin modifying enzymes[19].Second,the pleiotropic effect of current epidrugs leads to the combined inhibition of many members of a given family of epienzymes that have a broad spectrum of action and opposing roles in cancer cells.Third,and most importantly,cancer cell plasticity,and the switch between stem and non-stem state,is orchestrated by complex mechanisms,including epigenetic silencing of CSC markers and pluripotency genes.Despite genetic heterogeneity among cancer cells[52](due to stochastic or chemo-/radio-induced mutations along tumor evolution/treatment),DNA methylation and histone deacetylation seem to represent typical mechanisms involved in repressing stemness markers in non-stem cancer cells,as previously demonstrated for CD44,CD133,and CD166.Therefore,inhibiting DNMT and HDAC may result in increased expression of CSC markers[37,42,48]along with an increased stemness potential.Last,patients included in these clinical trials often present metastatic or advanced disease and are recruited regardless of the molecular subtypeof cancer.As aberrant DNA methylation is an early step of carcinogenesis,advanced disease may not be the relevant stage for treatments with DNMTi and HDACi.
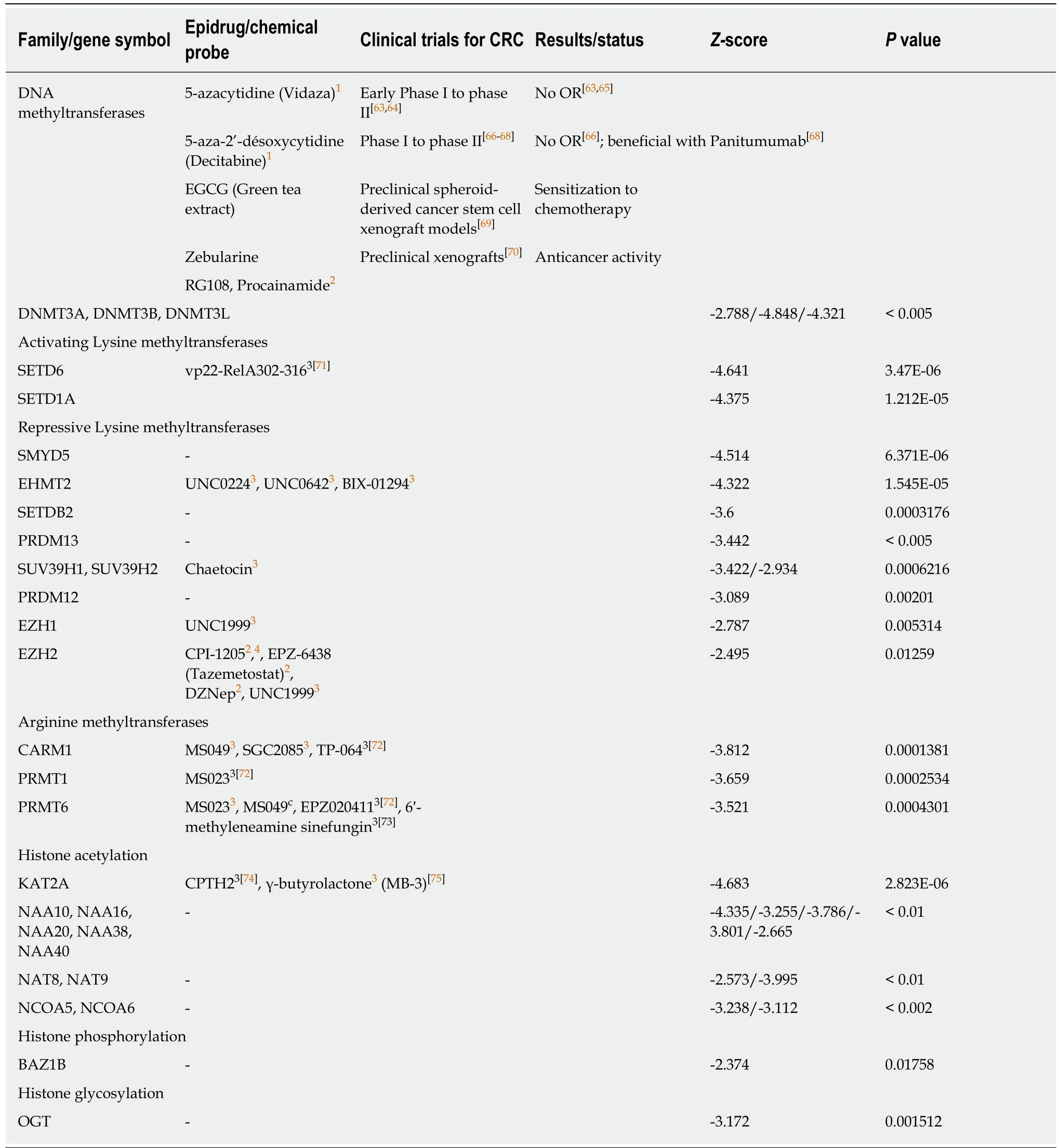
Table 1 Negative correlation between combined expression of cancer stem cell markers CD133,CD44 and CD166 and epigenetic writers
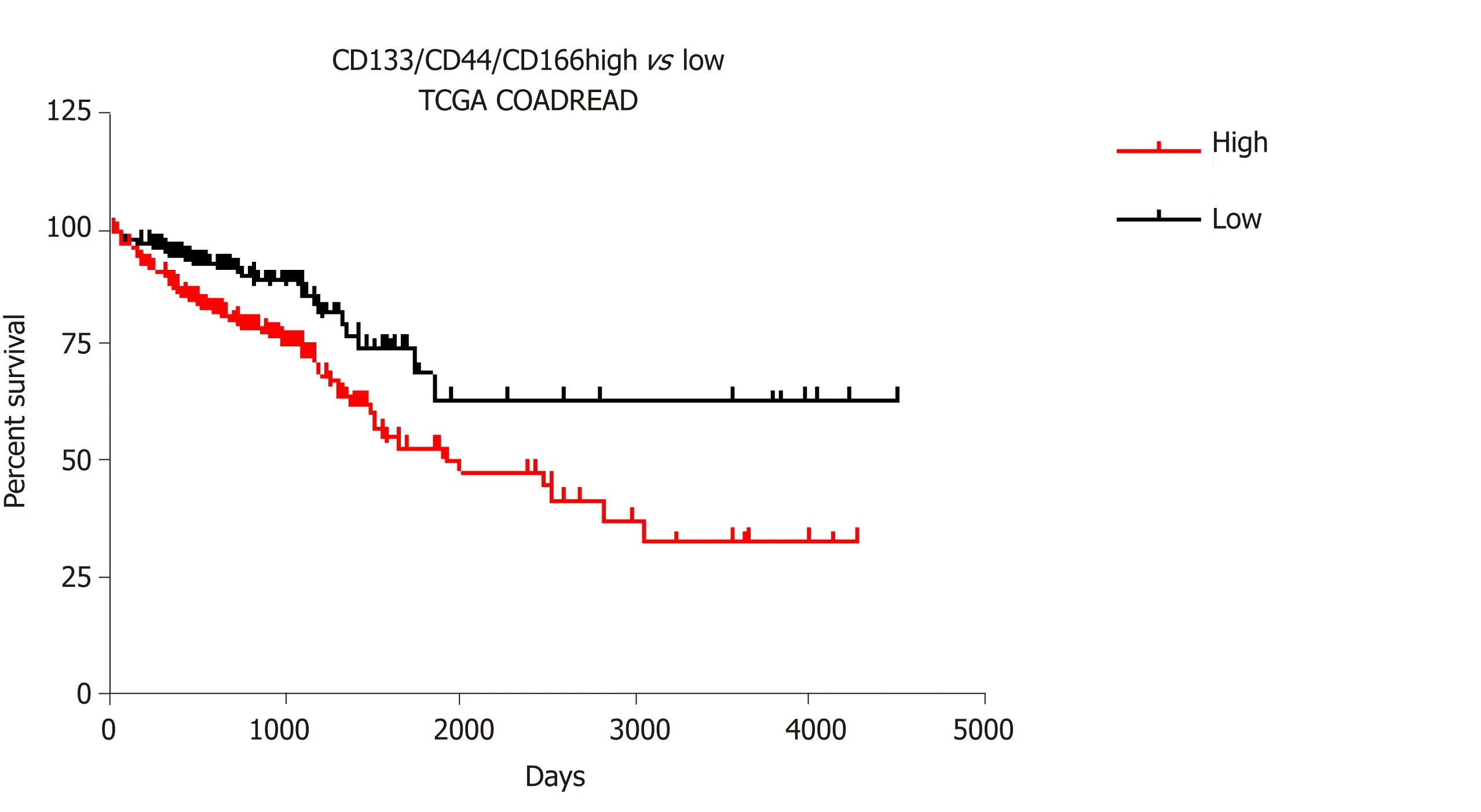
Figure 3 Survival analysis for CD133/CD44/CD166 expression profiles in colorectal cancer.
To refine these treatment strategies,tumor grade,heterogeneity,and subtypes of cancers will have to be considered.Indeed,determining which tumors will benefit from epigenetic differentiation strategies[53]and which tumors would acquire stemness capacities after epigenetic resetting is mandatory.Hence,modulating epigenetic alterations to sensitize cancer cells to other conventional therapies[54]or to lower their aggressiveness seems to be a reasonable goal when it comes to epigenetic strategies for advanced disease,as shown by numerous studies on cancer cell lines[53].HDAC and DNMT inhibitors,used alone or in combination,are able to sensitize resistant cancer cells and their use after conventional or targeted therapies have proven their efficacy in clinical trials[55].For instance,treatment with 5-azacitidine or 5-Aza-2’-deoxycitidine increases sensitivity of colon cancer cells to irinotecan and 5-FU[56].Irinotecan sensitivity with DNMTi was confirmed inin vivoCRC models showing tumor regression and increased survival in contrast with monotherapies.The same results were observed with the combination of 5-azacitidine and a BRAF inhibitor in CRC xenograft models[57].Synergetic therapies were also observed with HDACi in combination with 5-FU.Indeed,trichostatine A in combination with 5-FU suppresses colon cancer cell viability[58].However,initiating re-differentiation in CSCs remains a challenge dependent on the characteristics of each tumor type and with their specific genetic alterations.
Molecular subtypes of CRC or chemoresistance also predict how and whether or not patients will benefit from existing epidrug treatments.For instance,it has been shown that treatment with 5-azacitidine can restore chemosensitivity to irinotecan in microsatellite stable CRC cell lines but not in microsatellite instable CRC cell lines[56].Moreover,microsatellite instability CRC status is associated with the hypermethylation of glutathione peroxidase 3,a gene encoding an antioxidant selenoprotein involved in drug metabolism.In this case,treatment with 5-azacitidine induced an increase of glutathione peroxidase 3 expression and a decrease of chemosensitivity to oxaliplatin in microsatellite instability CRC cell lines[59].These findings emphasize the need for personalized therapies that consider CRC interindividual heterogeneity and classification.
Exploring new avenues for colon cancer treatment
In order to better anticipate how colon CSCs will respond to the different existing therapies,we analyzed TCGA_COADREAD data of 379 colon cancer patients using LinkedOmics[60].With this meta-analysis we assessed the correlation Z-score estimate(Stouffer method-based) and aPvalue between the combined expression of the three colon CSC markers CD133,CD44,and CD166 and an exhaustive list of known chromatin modifying enzymes (epigenetic writers and erasers) and chromatin binding proteins (epigenetic readers).The observed negative and positive correlation of expression between the three CSC markers and a significant number of epienzymes are highlighted in Tables 1 to 6.
Strikingly,DNMT3A,DNMT3B,and DNMT3L,the DNA methyltransferases that are responsible forde novoDNA methylation,showed a negative correlation score with the combined expression of the three CSC markers studied (Table 1),while the expression of DNMT1,responsible for DNA methylation maintenance,was not significantly correlated with the combination of these markers (-2 <score <2).Similarly,three class I and II HDAC as well as two sirtuins were found negatively correlated to the combination of markers (Table 2).None of the known HDAC were found positively correlated with the expression of the three CSC markers.This strongly suggests that inhibiting DNMT or HDAC activity would have no effect in colon cancers overexpressing CSC markers (and potentially harbor high stemness properties) but may have adverse effect in low-expressing and maybe less aggressive colon cancers.These data are in accordance with disappointing clinical trials that have been conducted so far with these inhibitors in colon cancer patients.Interestingly,our analyses suggest that another strategy to regulate DNA methylation in colon CSCs may be the inhibition of the methylcytosine dioxygenase TET2,known to trigger DNA demethylation and found correlated to CSC marker expression in our analyses(Table 3).
The correlation scores we obtained for other chromatin writers,readers,and erasers seem more specific to the enzyme itself than to their role in the shaping of epigenetic landscapes (Tables 1-6).
We found a negative correlation between the expression of the three markers and several histone lysine methyltransferases associated with the establishment of constitutive or facultative heterochromatin,including EZH2 that has recently emerged as one of the new favorite targets for epigenetic therapies[20](Table 1).These estimated scores in colon cancer expressing CD133,CD44,and CD166 suggest that an activator of EZH2,such as CPI-1205,may have better efficacy than known inhibitors in clinical trials to influence cancer stemness and are in accordance with a protective role of EZH2 in cell differentiation.Similarly,expression of EHMT2 (also known as G9A and KMT1C),encoding another lysine methyltransferase that also recently raised interests in the epidrug field,was inversely correlated with the three CSC markers expression (Table 1).
Only few lysine methyltransferases associated with gene activation were found correlated or inversely correlated with the combined expression of the three markers.Among them,SETD7 (Table 4),but not SETD6 (Table 1),may be a good candidate to inhibit stemness in colon cancer cells.
Recently,small molecules that can target specific bromodomains have been extensively developed[61].Bromodomains are part of a family of epigenetic readers that play pivotal roles in transcriptional regulation through the binding of acetylated histones and the recruitment of other epienzymes in epigenetic complexes at specific sites.We found only a few bromodomain-containing proteins whose expression was positively (BPTF,BAZ2B,Table 5) or negatively (BRD7,Table 6) correlated to the combined expression of the three CSC markers.
Among epigenetic readers,methylated DNA binding proteins have probably been overlooked as epidrug targets since expression of both MBD1 and MBD2 is positively correlated with CSC markers (Table 5).
Targeting members of the lysine-specific histone demethylase family of histone demethylases using inhibitors such as GSK-J1 may also be a good option since only a few of them are inversely correlated with the three CSC markers while KDM3B,KDM4B/C,KDM5B,KDM6A (UTX),and KDM6B (JMJD3) are positively correlated to their expression (Table 3).
Finally,JAK1/2 kinases,which possess a histone phosphorylation activity and are the targets of numerous inhibitors already tested in the clinic,mainly for other diseases,should probably be reconsidered for colon cancer patients with high expression of CD133,CD44,and CD166 or after conventional therapies.Indeed,our meta-analyses suggest that their expression is positively correlated to the expression of the three CSC markers (Table 4).
As mentioned above,CD44,CD133,and CD166 are potent markers of CSCs from multiple tissues including digestive (gastric,pancreatic) and non-digestive cancers in which they are epigenetically regulated.Therefore,these considerations could be largely applicable to other types of cancers,in which correlation studies between epienzymes and CSC markers may be of great interest.
PERSPECTIVES
Although epigenetic therapies are conceptually very promising,several pitfalls will have to be overcome in order to take a step forward in clinical trials for solid tumors.First,while intra-tumor and inter-individual heterogeneity of CRC is now evident,epigenetic landscapes and epienzyme activity will have to be studied in all types of tumor cells.Single cell approaches will be very useful to circumvent the difficulty of exploring rare CSCs from different CRC consensus molecular subtypes.Second,studies to prove causal correlations between epienzyme expression and the control of stemness will be mandatory in order to clear up confusion relative to the oncogenic or tumor suppressive roles of chromatin modifiers.Finally,the major difficulty for the design of new epidrugs is to target efficiently a single member of entire families of epienzymes that have homologous domains but different roles in stemness.To circumvent this difficulty,increasing specificity by targeting epigenetic complexesand therefore epienzyme-epienzyme interactions may be a better option for new designs.Based on these considerations,epigenetic personalized medicine will be truly envisioned.
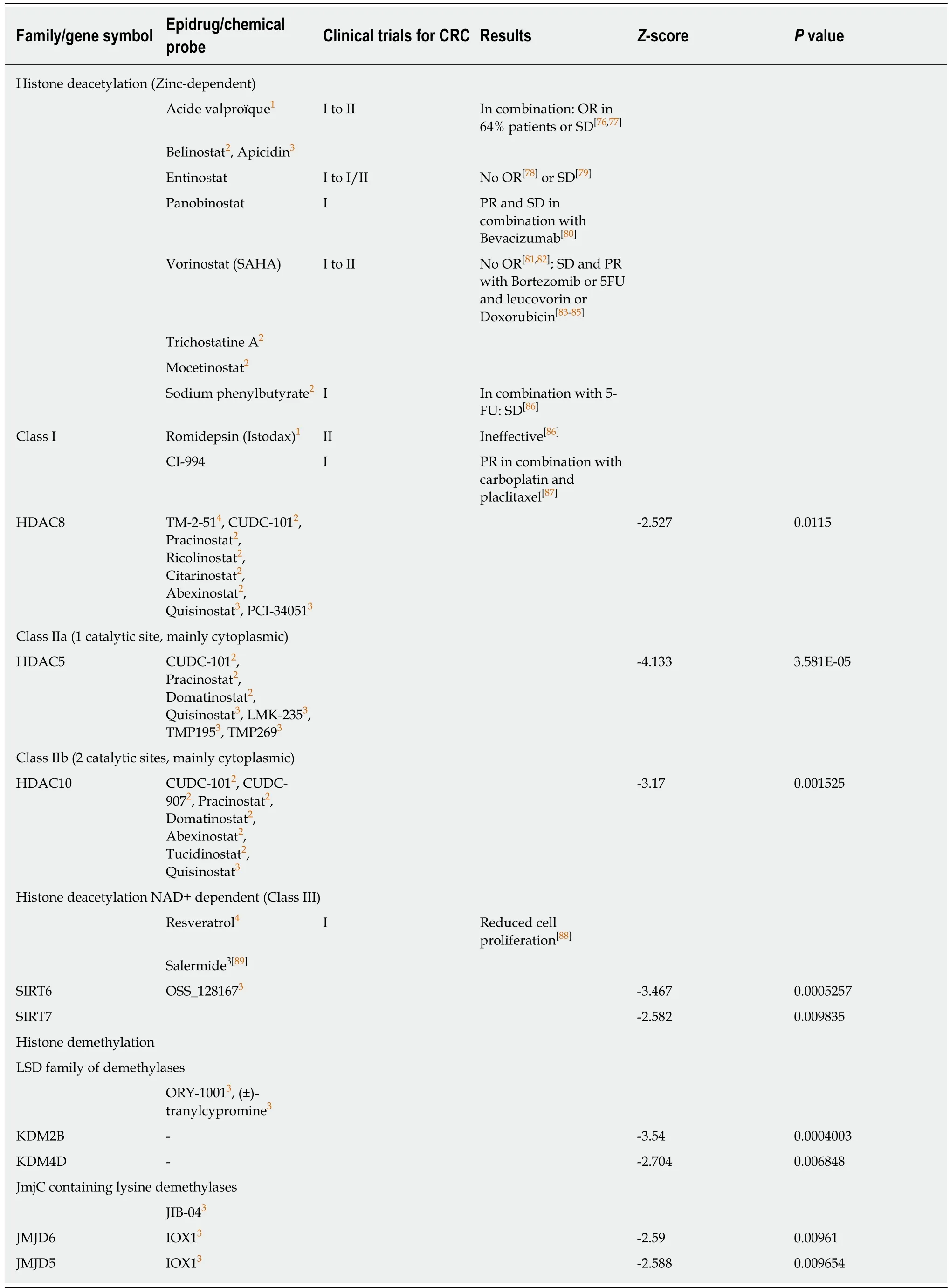
Table 2 Negative correlation between combined expression of cancer stem cell markers CD133,CD44 and CD166 and epigenetic erasers

Table 3 Positive correlation between combined expression of cancer stem cell markers CD133,CD44 and CD166 and epigenetic erasers
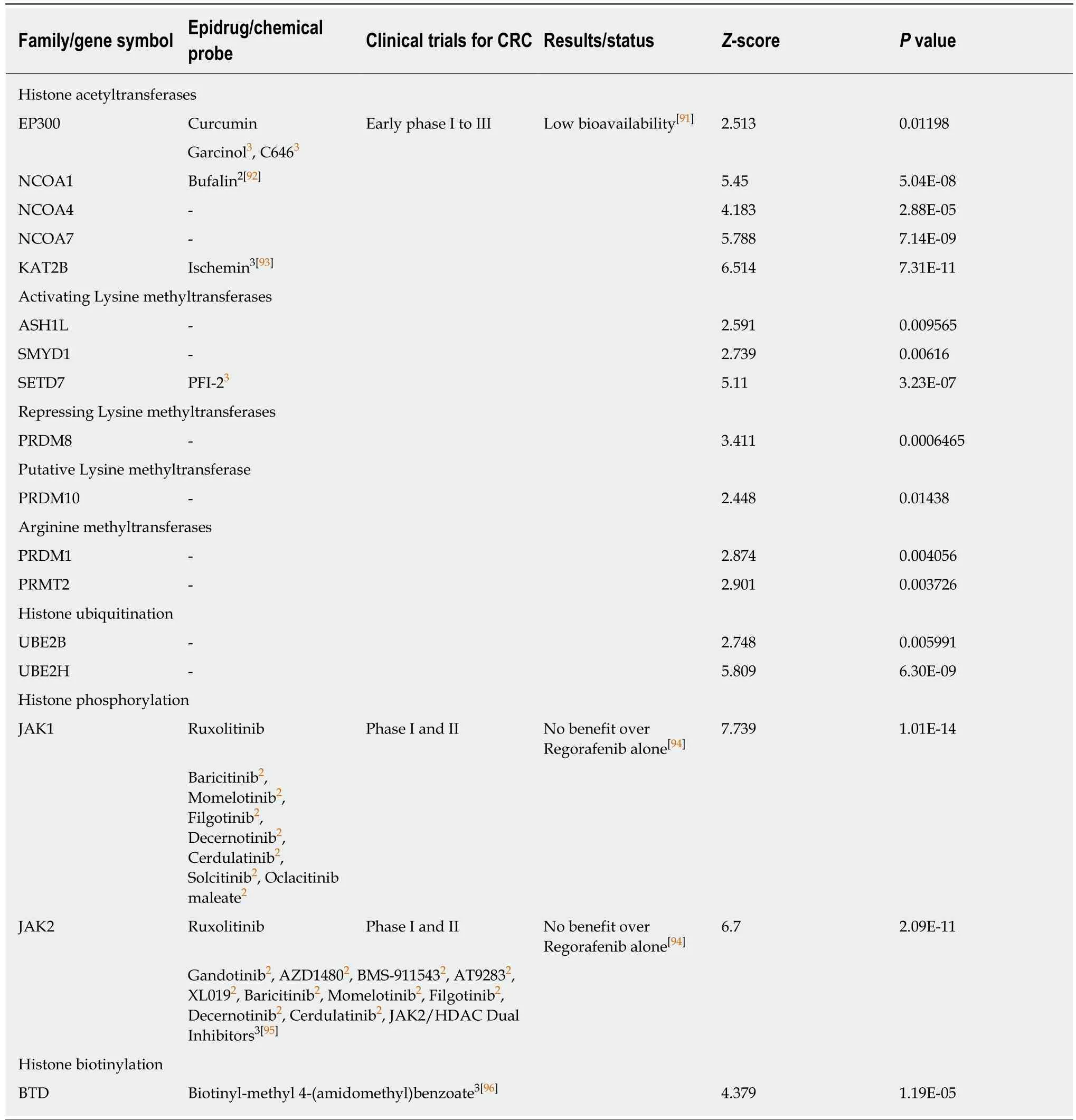
Table 4 Positive correlation between combined expression of cancer stem cell markers CD133,CD44 and CD166 and epigenetic writers
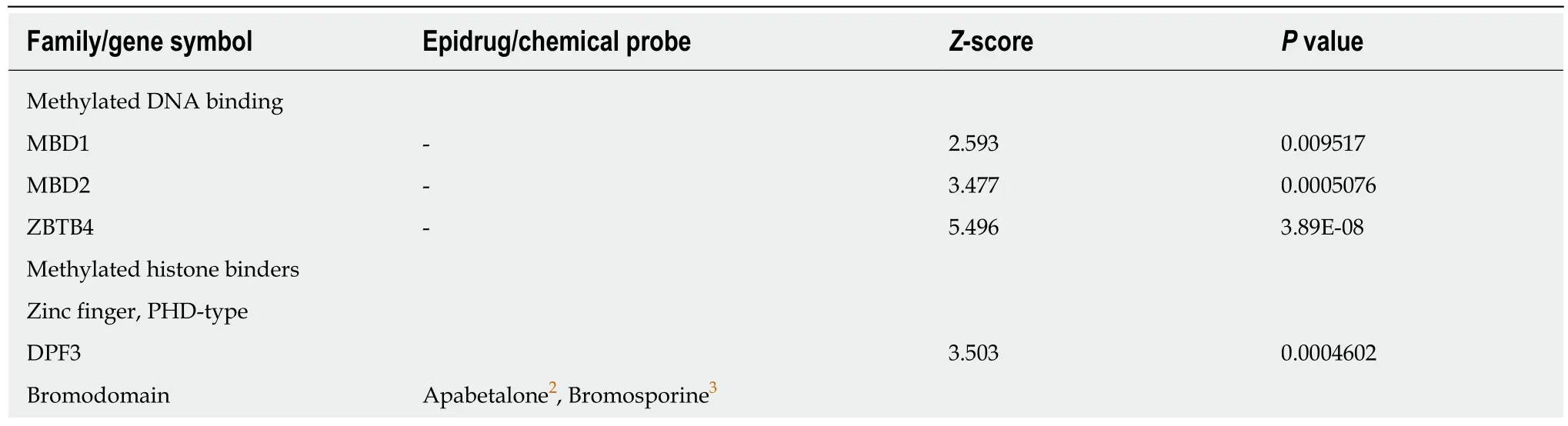
Table 5 Positive correlation between combined expression of cancer stem cell markers CD133,CD44 and CD166 and epigenetic readers

1Approved for the treatment of other diseases;2Used in clinical trials for other diseases;3Not yet used in clinical trials.
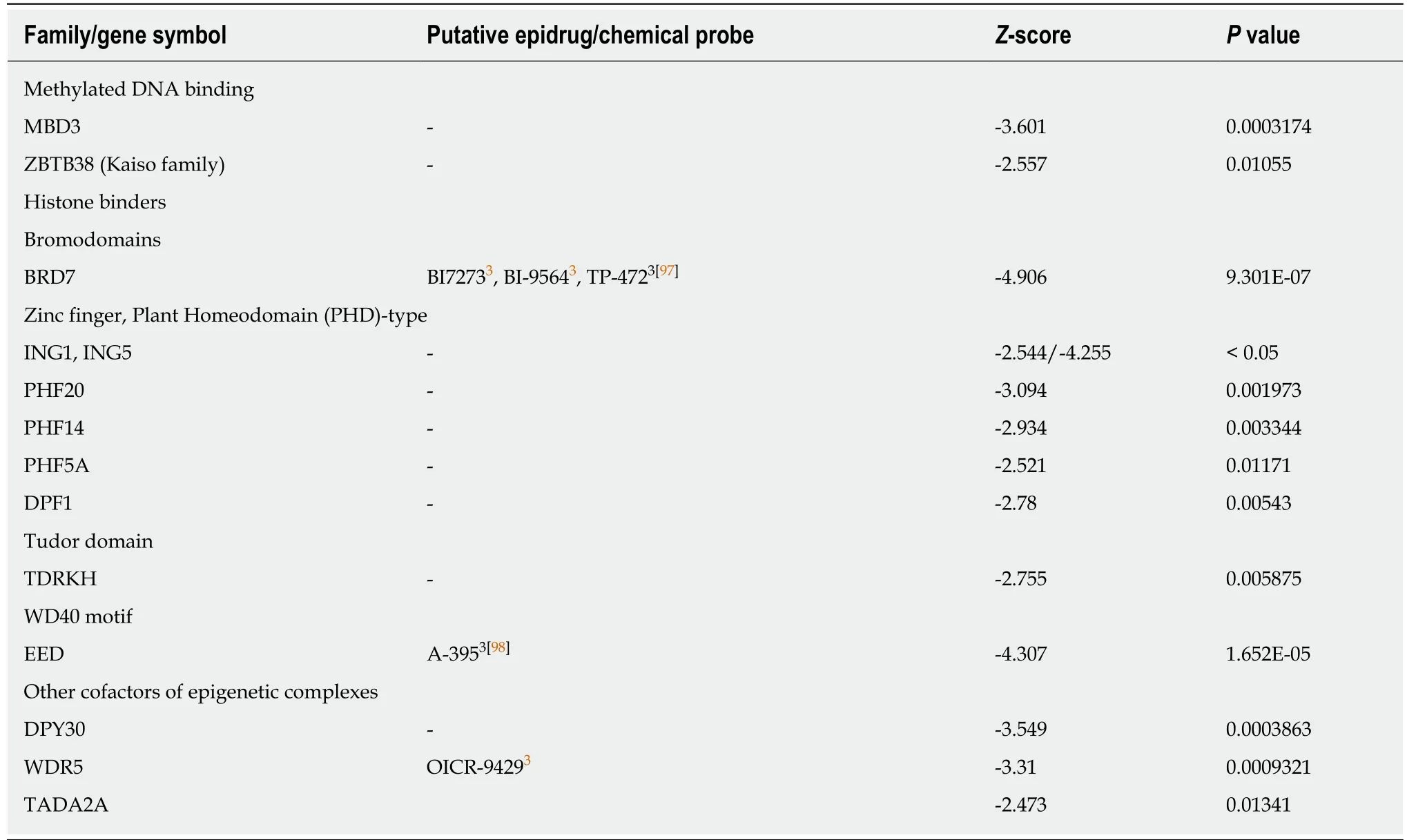
Table 6 Negative correlation between combined expression of cancer stem cell markers CD133,CD44 and CD166 and epigenetic readers
ACKNOWLEDGEMENTS
The authors would like to thank Dr Samuel Malone for his careful and critical reading of the manuscript,for the helpful comments and for English editing.
杂志排行

World Journal of Stem Cells的其它文章
- Unexpected encounter of the parasitic kind
- CRISPR/Cas system: An emerging technology in stem cell research
- Cytokine interplay among the diseased retina,inflammatory cells and mesenchymal stem cells - a clue to stem cell-based therapy
- Developments in cell culture systems for human pluripotent stem cells
- Monitoring maturation of neural stem cell grafts within a host microenvironment
- Ameliorating liver fibrosis in an animal model using the secretome released from miR-122-transfected adipose-derived stem cells