利用加权基因共表达网络分析构建食管腺癌预后枢纽基因网络
2019-11-16陈超童国俊张建斌沈亮何焕钟
陈超 童国俊 张建斌 沈亮 何焕钟
食管癌是因癌症导致死亡的第六大原因,5年生存率仅为19%,其中晚期食管癌患者5年生存率仅0.9%[1]。近年来,食管腺癌发病率逐年上升;在西方国家,食管腺癌已成为发病率最高的食管恶性肿瘤[2]。随着医疗水平的发展,食管腺癌患者的预后改善仍十分有限。在复杂的肿瘤微环境下,传统的单基因研究因不能揭示肿瘤基因复杂的信号传导网络而存在很大的局限性。随着基因芯片、转录组测序(RNA-seq)技术的发展,应用生物信息分析实现多个基因表达及功能研究的方法,为疾病发生、发展的机制探索提供了新思路。本研究提取癌症和肿瘤基因图谱计划(TCGA)数据库中78例具有RNA-seq的食管腺癌标本信息,通过加权基因共表达网络分析(weighted gene co-expression network analysis,WGC原NA)研究肿瘤的RNA-seq数据,筛选与预后相关的模块及枢纽基因,并根据表达谱信息构建多个枢纽基因的共表达网络关系。现将结果报道如下。
1 资料和方法
1.1 数据来源与预处理 9例正常食管组织和78例食管腺癌组织的基因表达数据及临床预后数据均来源于免费、开源的TCGA数据库(https://cancergenome.nih.gov/)。将所有样本的基因名转化为标准基因名;数据的标准化处理是以正常食管组织为参照,对肿瘤标本数据进行归一化处理。
1.2 共表达网络的构建和模块的识别 利用R-3.5.1软件运行“WGCNA”包。为降低运算量,笔者筛选出基因表达量方差大于所有方差四分位数的基因用于共表达网络的构建。采用样本聚类树方法,根据聚类图剔除离群样本来保证构建稳定的共表达网络。构建无尺度网络使基因共表达网络符合无尺度现象,以无尺度网络指数(R2)=0.9作为满足无尺度条件的标准,同时根据平均连接度确定软阈值(茁)。利用拓扑重叠(TOM)矩阵、相异常度矩阵计算基因与基因间的关联程度[3];对基因构建层次聚类树图形,采用动态剪枝法计算基因模块颜色。计算基因模块的特征值(ME),引入临床信息,对ME进行分层聚类并绘制树状图,设置高度值0.25为分割线,合并相似程度较高的基因模块,再用剪切后的模块绘制新的聚类树和模块图。
1.3 表观数据的纳入和枢纽基因的筛选 读取、清洗临床样本表观数据,主要包括性别、年龄、肿瘤级别、生存时间等,将样本与表达矩阵进行匹配重建样本聚类树。计算模块与表观数据相关性,绘制模块内基因表达热图,根据生存时间性状与模块特征向量基因的相关性及P值来挖掘与该性状相关的模块。计算相关模块内基因显著性(GS)以及基因在模块内的模块隶属度(MM),设置枢纽模块中候选枢纽基因3个筛选标准,P.GS<0.05。同时计算加权基因共表达网络的权重值,筛选出权重值前200位的基因,与上述候选枢纽基因的交集为共同枢纽基因。
1.4 枢纽基因及共表达网络可视化 将共同枢纽基因及基因共表达网络的权重信息导入Cytoscape3.7.0软件,根据互作网络关系绘制枢纽基因共表达网络图。
2 结果
2.1 TCGA数据库中食管腺癌病例基本信息 本研究共纳入食管腺癌患者78例,其中男67例,女11例;年龄 28.0~86.6[68.4(58.0,77.1)]岁;生存时间(634依513)d;随访时间(603依524)d;国际抗癌联盟食管癌TNM分期:玉期10例,域A期9例,域B期16例,芋期33例,郁期10例。
2.2 共表达网络的构建与枢纽模块的选择 基因表达量方差大于所有方差四分位数的基因共9 933个,通过聚类树删除离群样本14例,剩余64例食管腺癌样本纳入下一步分析。根据R2=0.9、茁=5作为标准,当茁=5时,无尺度拓扑网络的R2=0.95,见图1。引入生存时间信息,采用动态剪切法将树剪切成不同的模块,合并相似度较高的模块,见图2a(插页)。通过模块样本性状相关性热图,发现深蓝色模块与生存预后密切相关,相关系数(Cor)=0.3,P=0.01,即深蓝色模块为枢纽模块,见图2b(插页)。枢纽模块中基因内部连接度与MM相关,深蓝色模块与基因显著性相关,Cor=0.61,P<0.01,见图 3。

图1 WGCNA的茁确定(a:不同茁下计算的无尺度网络符合指数;b:不同茁下计算的平均连接度;c:茁=5时连接度分布直方图;d:茁=5时无尺度网络拓扑检测)
2.3 枢纽基因确定及共表达网络可视化 在深蓝色枢纽模块中,根据的标准,筛选得到20个候选枢纽基因集A,再根据基因权重共表达网络的权重大小筛选得到32个候选枢纽基因集B,基因集A和B的交集基因有19个,见表1。将19个基因间的权重信息导入Cytoscape软件后得到基因间互作网络图,其中基因间共表达权重系数最大的3对基因分别是FOLH1和SCRG1、FOLH1和UGT2B15、FOLH1和SFTB,见图4。

图2 动态剪切树与模块样本性状相关性热图(a:确定β=5,合并相似度较高的模块;b:引入生存时间作为样本性状,显示与样本性状相关的模块)

图3 深蓝色模块中GS与MM的关系(右上角为的基因分布)
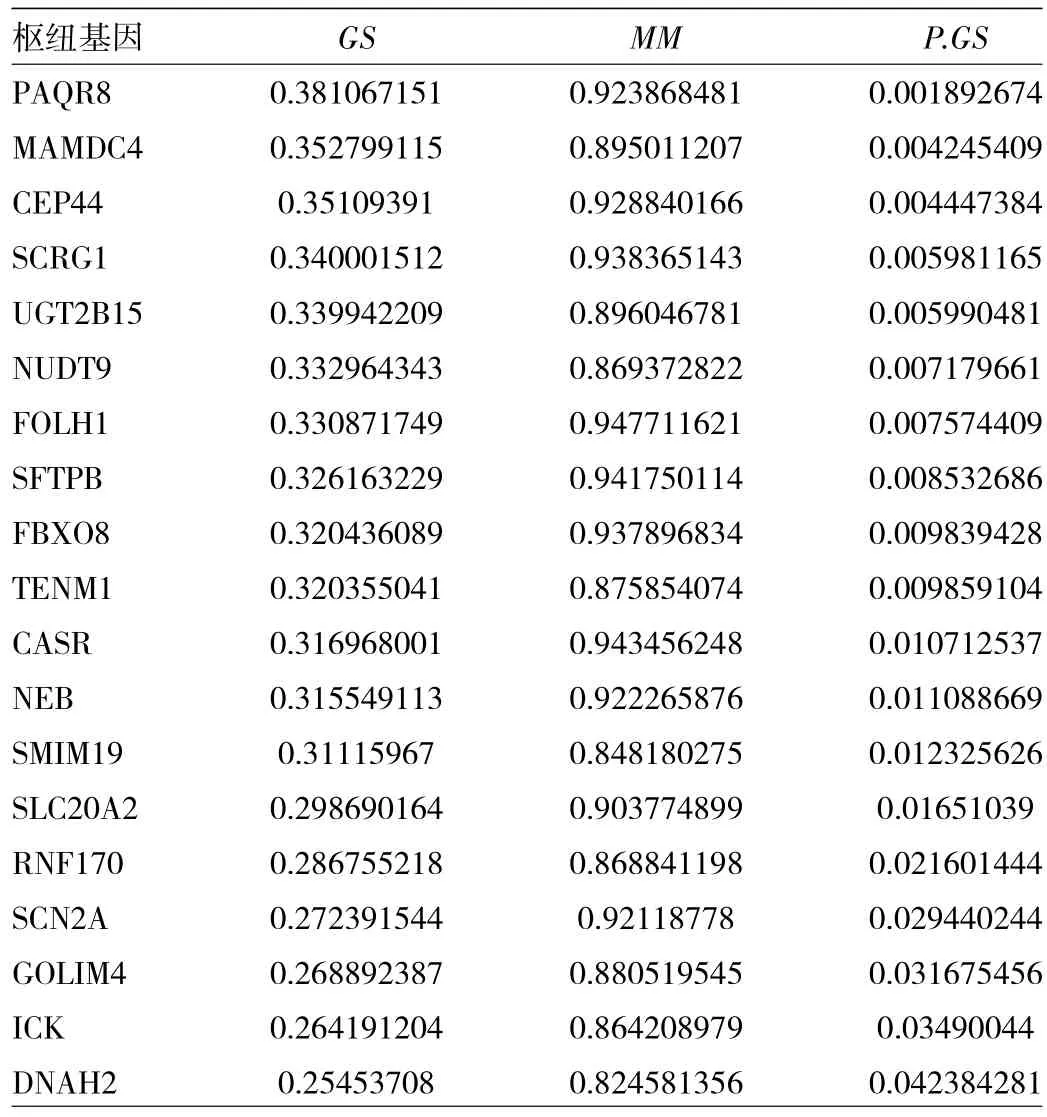
表1 19个与临床预后相关的枢纽基因GS、MM和P.GS

图4 枢纽基因之间的共表达网络关系图
3 讨论
胃食管反流、吸烟、肥胖、饮食习惯是食管腺癌的危险因素[2]。而在危险因素影响下的表观遗传异常修饰导致基因转录水平改变是肿瘤预后研究的基础。局限于某个基因或某几个分子的传统研究方法不能全面地阐述肿瘤的发展及预后。WGCNA在2005年首先被提出,是指根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点[4]。相比于传统的单基因研究,WGCNA利用数千或近万个变化最大的基因或全部基因的信息识别感兴趣的基因模块,并与表型进行显著性关联分析。该方法创新性强,研究结果可信度较高。近期亦有不少期刊发表了利用WGCNA的相关研究[5-7]。
本研究利用WGCNA处理78例食管腺癌标本基因的RNA-seq数据及临床数据,通过深层次挖掘得到19个枢纽基因,并构建了枢纽基因的共表达网络。WGCNA构建的预后相关的枢纽基因共表达网络图中,FOLH1与SCRG1、UGT2B15、SFTB等3个基因的共表达权重系数最高,属于共表达网络图中的核心基因。FOLH1是一种域型跨膜糖蛋白,其分子量约为100kDa,由750个氨基酸组成[8]。FOLH1主要在前列腺、中枢及外周神经系统、肾脏、小肠和肿瘤相关的新血管系统中表达[9],可通过谷氨酸代谢来调节叶酸的吸收[10]。有研究认为FOLH1表达水平能评估肿瘤患者的预后,尤其在已发生肿瘤转移的情况下;同时发现经普通筛查发现的恶性肿瘤分级与FOLH1表达亦密切相关[11]。Chang等[12]称FOLH1在多种恶性肿瘤的新生血管系统中均有表达,可作为抗肿瘤新生血管生成治疗的有效靶点。目前关于FOLH1在前列腺肿瘤中表达的研究较多[13-15],亦有FOLH1与乳腺癌[16]、非小细胞肺癌[17]等肿瘤预后有关的报道。然而,关于FOLH1在食管恶性肿瘤中表达的研究尚未见报道。根据WGCNA构建的共表达网络分析以及关于FOLH1的现有研究结果,笔者认为FOLH1可能是食管腺癌中新的预后相关分子。
本研究通过WGCNA构建加权基因共表达网络,初步筛选得到与食管腺癌预后相关的19个枢纽基因及其共表达网络关系,为食管腺癌的治疗提供新靶点。
