配体保护下Znm Cdn Se10(m + n =10)量子点的密度泛函理论研究
2019-11-13余盛萍黄兴隆范滔滔
余盛萍,黄兴隆,范滔滔
(1.西南民族大学化学与环境保护工程学院,四川 成都 610041;2.四川大学原子与分子物理研究所,四川 成都 610065)
量子点(QDs),即零维纳米材料,是一种新型的Ⅱ-Ⅵ族或Ⅲ-Ⅴ族元素组成的半导体纳米粒子(NPs),又称半导体纳米晶体(NCs),在发光材料、光敏传感器、生物成像和太阳能电池等领域[1-13]具有广泛应用.CdSe 量子点虽然具有激发光谱宽且连续分布、发射光谱窄且对称、波长可调、光化学稳定性高、荧光寿命长等优点,但由于CdSe 量子点的生物毒性使其在生物检测方面的应用受到限制[5-6].而ZnS、ZnSe 和ZnTe 等量子点由于带隙宽且能够克服此缺点而被广泛关注[7-13].为了促进半导体量子点在实际生产中的应用,在合成量子点时将Zn 引入到CdSe 量子点的晶格中,形成三元合金量子点.目前已有关于Zn 掺杂到CdSe 量子点的研究报道[14-15],引入PH3配体会阻碍QDs 晶粒相互间发生聚集,起到了稳定剂的作用[16].但对于PH3配体对其ZnmCdnSe10吸附的理论研究还比较缺乏.本论文采用密度泛函方法系统的研究了生物毒性更低的ZnmCdnSe10三元量子点与PH3配体之间的相互作用方式及其吸附产物的结构和性质,为设计高品质量子点奠定基础.
1 计算方法
本文用不同数量的Zn 原子逐渐替代Cd10Se10中的不同位点上的Cd 原子,形成ZnmCdnSe10(m+n =10)三元量子点,在TPSS/def2-TZVP 水平下优化其结构,得到其最稳定构型并比较他们的各种性质.然后将PH3配体吸附到ZnmCdnSe10上,并研究其吸附产物的结构和性质.电子与电子之间的相互作用中的交换相关效应通过广义梯度近似(GGA)进行校正,采用超软赝势来描述离子实与价电子之间的相互作用势,用GGA 中的TPSS[17]来处理电子之间的交换关联能,基组为def2-TZVP[18].为了验证此方法的可靠性,计算了Zn6Se6的Zn-Se 的键长为2.411 Å,与文献报道的2.382 Å[15]比较吻合.所有计算工作使用Turbomole 软件包完成.
2 结果与讨论
2.1 ZnmCdnSe10(m +n =10)的结构
图1 列出了三元团簇ZnmCdnSe10(m+n=10)的最稳定构型.左边键长为Se-Cd 平均键长,在2.572 Å ~2.634 Å 之间变化;右边键长为Se-Zn 键平均键长,其变化范围为2.243 Å ~2.459 Å.Se-Cd 平均键长比Se-Zn 键平均键长长,这是由于Cd 原子半径比Zn 原子半径小的原因.如图1 所示,除Cd5Zn5Se10外,其它最稳定的三元量子点结构中的Zn 原子优先占据α位,其次再占据β位,最后占据 γ位.这是因为α位的活性更高更容易掺杂入Zn 原子.
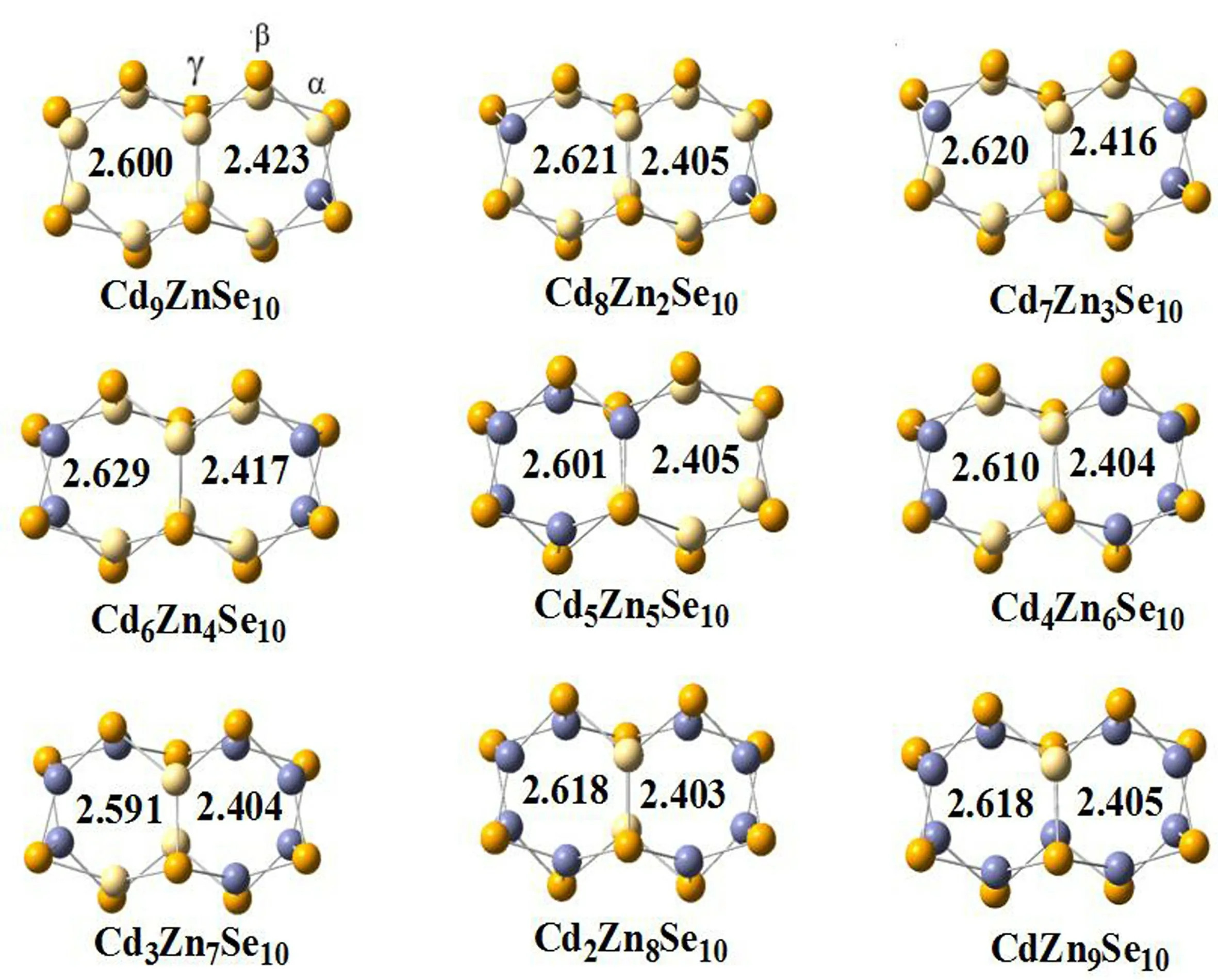
图1 在TPSS/def2-TZVP 水平下优化得到的团簇ZnmCdnSe10(m+n =10)的最稳定构型(键长单位为:Å)Fig.1 The most stable Znm,CdnSe10(m+n =10)clusters at the TPSS/def2-TZVPlevel(All bond lengths are in Å)
2.2 配体PH3在ZnmCdnSe10(m+n =10)上的吸附
图2 给出了ZnmCdnSe10(PH3)10(m+n=10)吸附产物的最稳定构型,对比图1 和图2 可知,除Zn 原子个数n =5,6 和7 外,最稳定的“裸”三元团簇和配体保护的三元团簇的Zn 原子位置一致,说明配体对团簇影响在引入5,6 和7 个Zn 原子时较大.表1列出了PH3的平均吸附能.吸附上配体后,掺杂量子点原子时影响较大.表1 列出了PH3的平均吸附能.吸附上配体后,掺杂量子点的结构变化不大,其Zn-Se 平均键长变化也不大.随着掺杂原子的增多,平均吸附能从0.27 eV 逐渐增加到0.32 eV,配体的加入,阻碍了量子点团簇的聚集.新生成的配位键P-Zn 键和P-Cd 键的键长分别约为2.400 Å 和2.900 Å.

图2 TPSS/def2-TZVP 水平下优化得到ZnmCdnSe10(PH3)10(m+n =10)团簇的最稳定构型(键长单位为:Å)Fig.2 The most stableZnmCdnSe10(PH3)10(m+n =10)clusters at the TPSS/def2-TZVPlevel(All bond lengths are in Å)

表1 在TPSS/def2-TZVP 水平下优化的复合物ZnmCdnSe10(PH3)10(m +n =10)的吸附能,净电荷和HOMO-LUMO 能隙Δε(eV).Table 1 The adsorption energy(Eadv,in eV),net charge and HOMO-LUMO gap(Δε,eV)of the most stable ZnmCdnSe10(PH3)10(m +n =10).
2.3 HOMO 和LUMO 轨道
图3 列出了最稳定结构的LUMO 轨道.LUMO 轨道主要分布在ZnmCdnSe10(m+n =10)上,集中到中间两环交界处.从LUMO 图可以看出,每个团簇的LOMO 均表现出一定的对称性,这说明配体于空间的位阻和对称性同样影响着整个团簇的LOMO.表1 同时列出了HOMO-LUMO 能隙值,HOMO-LUMO 能隙值的大小反应了电子从占据轨道向空轨道跃迁能力的大小,因此在一定程度上代表了分子参与化学反应能量的强弱.从表中可以看出,随着掺杂Zn 原子的数目增多,Δε的值越来大.说明电子从占据轨道向空轨道跃迁更困难,也就是掺杂原子越多,团簇的稳定性越高.

图3 TPSS/def2-TZVP 水平下优化得到的最稳定ZnmCdnSe10(PH3)10(m+n =10)的LUMO 轨道Fig.3 LUMO orbitals of the most stableZnmCdnSe10(PH3)10(m+n =10)clusters at the TPSS/def2-TZVP level
2.4 电荷分析
表1 给出所有吸附产物的最稳定结构的净电荷值.我们可以看出配体为正电荷,ZnmCdnSe10为负电荷,表明有电子从PH3配体转移到团簇ZnmCdnSe10上,团簇为电子受体,配体为电子供体.PH3配体的作用吸附在团簇ZnmCdnSe10表面,不仅阻碍团簇聚集,而且部分地改变了ZnmCdnSe10的电子结构.当PH3配体吸附到团簇上时,团簇ZnmCdnSe10的电荷随着掺杂Zn 原子增多变化不大,即其净电荷变化不大,约为-0.41 e.
2.5 态密度
图4 中绘出了最稳定结构吸附产物ZnmCdnSe10(PH3)10(m+n =10)的态密度,虚线表示Fermi 能级,红色为s 轨道,蓝色为p 轨道,橄榄绿色为d 轨道.在Fermi 能级附近,s,p,d 轨道简并,轨道杂化率较高.在-6 到6 之间,p 轨道主导.
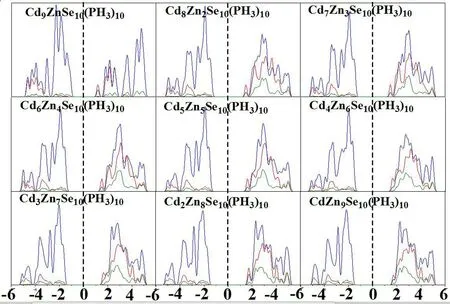
图4 最稳定ZnmCdnSe10(PH3)10(m+n=10)团簇的态密度(红、蓝、绿线分别表示s、p、d 态)Fig.4 Projected density of states(PDOS)for the most stable ZnmCdnSe10(PH3)10(m+n =10)cluster.Fermi level is set at zero on the energy axis(red:s states;blue:p states;Green:d states)
3 小结
在TPSS/def2-TZVP 水平下,优化得到了ZnmCdnSe10团簇的稳定构型,然后PH3配体吸附到ZnmCdnSe10团簇上,研究其配体效应,结果表明:第一,吸附能随着掺杂Zn 原子的增多而略微增大;第二,LUMO 轨道都主要占据在ZnmCdnSe10上,量子点的HOMO-LUMO 能隙随着掺杂Zn 原子的增多而增大;第三,电子从PH3配体流向ZnmCdnSe10主体,PH3是电子供体,ZnmCdnSe10是电子受体,表明PH3配体的存在不仅阻碍了掺杂量子点ZnmCdnSe10的聚集,而且还部分改变了掺杂量子点ZnmCdnSe10的电子结构,电荷转移变化不大;第四,各个吸附产物的态密度中p 轨道占主导.
