钙通道抑制介导轴突稳定对视神经损伤修复作用的研究进展
2019-11-12喻哲昊王鹏飞
喻哲昊,王鹏飞,曹 霞
0引言
外伤性视神经损伤是导致不可逆失明的主要原因,并且可造成因视神经轴突变性和视网膜神经节细胞(RGCs)凋亡而导致的进行性视力损害。迄今为止,无论是药物治疗还是手术治疗都没有很好的疗效,无法阻止视力丢失。轴突再生是视神经损伤后视觉功能恢复的关键,视神经损伤后,RGCs轴突通常不能再生,进而导致视觉功能丧失。轴突的再生对于神经连接的恢复以及损伤后视觉系统功能的重建至关重要。与其他中枢神经系统轴突相似,视神经具有非常有限的再生能力。到目前为止,还没有有效的治疗方法促进轴突向远端再生,修复视觉通路的轴突连接[1]。
最初的损伤导致钙迅速流入轴突,激活钙蛋白酶系统、c-Jun N-末端激酶(JNK)系统以及Akt信号途径来发挥促凋亡功能,造成轴突变性和RGC凋亡。已有研究表明,引发急性轴突变性(AAD)的主要事件之一是轴突内钙的流入,而当钙离子流入被抑制时可显著减轻轴突变性[2]。本文通过研究钙通道抑制剂对轴突变性、RGCs存活和轴突再生的影响, 对钙通道抑制与视神经损伤修复方面的研究和进展做一综述。
1钙蛋白酶途径
1.1钙蛋白酶钙蛋白酶是细胞内钙浓度依赖型的中性半胱氨酸内肽酶。在胞内,钙蛋白酶活性通过钙离子浓度增加而增强。钙蛋白酶可以裂解各种细胞成分,主要作用于细胞骨架蛋白、蛋白激酶和磷酸酶,还参与细胞内的信号传递。轴突内低水平的Ca2+浓度由Ca2+-ATP酶和Na+/Ca2+泵来维持,两者都直接或间接地依赖于一个恒定的能量供给。损伤影响轴突能量生成和供给,导致轴突内钙离子逐渐积累,最终会激活钙蛋白酶系统[3]。Zhang等[4]通过研究发现,视神经发生AAD的早期,钙离子迅速流入轴突内,使轴突内钙离子浓度增加,从而激活钙蛋白酶,导致细胞骨架降解。
1.2钙蛋白酶抑制细胞内游离钙是钙蛋白酶活化的主要调节剂,但钙蛋白酶也通过与特异性抑制剂——钙蛋白酶抑素相互作用来调节,钙蛋白酶和钙蛋白酶抑素在中枢神经系统(central nervous system,CNS)中的所有细胞类型中表达[5]。钙蛋白酶抑素(calpastatin)是一种由钙蛋白酶抑制蛋白(CAST)基因编码的蛋白质,该基因编码的蛋白质是一种内源性钙蛋白酶抑制剂,为轴突变性的主要内生调控器。钙蛋白酶抑素是唯一已知的蛋白酶抑制剂,目前研究表明其专一性地抑制钙蛋白酶活性,可抑制初始活化和活化后期的钙蛋白酶活性[6]。钙蛋白酶抑素在神经系统内被广泛表达。研究表明物理损伤后,在退化的轴突上发现钙蛋白酶抑素的消耗,从而影响了轴突的存活水平。轴突变性不仅发生在病理条件下,而且还发生在正常神经发育过程。在视神经内,钙蛋白酶抑素调节正常RGCs轴突的重塑。Yang等[7]通过将对照scrambled-shRNA和calpastatin-shRNA质粒电穿孔到野生型小鼠的RGC中来确定内源钙蛋白酶抑素在轴突重塑中的功能,发现calpastatin-shRNA靶向的绝大多数轴突已经发生重塑,表明内源性钙蛋白酶抑素在体内自主调节发育轴突变性。钙蛋白酶抑素亦可调节轴突内的沃勒氏变性。Ma等[8]在视神经轴突中表达人钙蛋白酶抑制素(hCAST)(内源性钙蛋白酶抑制剂)的转基因小鼠上研究了钙蛋白酶抑素与Wallerian变性的关系,发现在视神经横断后5d,hCAST转基因小鼠相较于对照小鼠其视神经丝蛋白水解降低且神经-肌肉连接的形态完整性有所提高。这些结果提供了直接证据表明钙蛋白酶抑制对轴突Wallerian变性有积极的影响。同时,为了测试内源性钙蛋白酶抑素在体内Wallerian变性中是否具有调节作用,对视神经横断后轴突中的蛋白水平进行检测,发现钙蛋白酶抑素有显著的消耗。这些发现表明,在损伤和发育过程中,内源性钙蛋白酶抑素是在损伤诱导和发育性退化过程中轴突生存的关键调节剂[7]。近期有研究证实, 拉坦前列素,一种治疗眼内压增加的药物,可以减弱轴突变性后的钙蛋白酶抑素的分解,从而保护RGCs免受创伤后神经元变性的影响[9]。
1.3钙蛋白酶抑制对视神经损伤的影响钙蛋白酶的抑制减弱了髓磷脂和轴突蛋白的损失,减少轴突变性[10]。钙蛋白酶抑制也可保证少突胶质细胞存活率,防止轴突神经纤维蛋白的退化,改善视网膜上RGC的存活率[11]。近年来,对于钙蛋白酶抑制在视神经损伤方面的研究一直在继续。在MAPK级联反应上游的衔接蛋白Sarm1是激活该MAPK级联所必需的。Sarm1-MAPK途径触发局部能量缺失,导致轴突退化变性。Yang团队通过使用钙蛋白酶抑制剂在切断轴突2h后处理轴突发现,该方法强烈抑制钙蛋白酶的活化和轴突变性,但均未改善受损轴突的能量缺乏。该实验表明钙蛋白酶在ATP消耗的下游发挥功能,轴突中的局部能量缺乏先于钙蛋白酶激活和损伤后的形态变性,并且是其原因[3]。Ma等[12]通过使用病毒载体介导的RGC转导,能够将特异性内源性钙蛋白酶抑素递送至最大损伤部位(前弯曲视神经轴突),从而使应变诱导的轴突内钙蛋白酶活性降低,减轻逆行运输的直接损害。经研究证实,小鼠的钙蛋白酶激活加速了轴突损伤和RGCs的凋亡,应用钙蛋白酶抑制剂可以减弱这一过程,结果表明钙蛋白酶抑制剂对轴突变性引起的RGC死亡具有保护作用。这条途径可能是预防损伤后轴突退化变性的重要治疗手段[13]。钙通道的抑制通过阻断钙离子内流而降低损伤视神经的钙蛋白酶活性,因此钙通道抑制减少钙蛋白酶的活性可以为轴突变性和RGC存活提供一种机械的联系[14]。
2 JNK/c-Jun途径
2.1 JNK/c-Jun信号通路的作用JNK是一种丝氨酸/苏氨酸激酶,JNK信号是一种被激活的蛋白质激酶(MAPK)家族的一个组成部分。Eph/ephrin信号通路涉及调节胚胎发育至关重要的一系列过程,研究发现ephrin A5与EphB2的相互作用导致JNK的持续激活,其控制细胞凋亡和细胞增殖的平衡,这是在眼睛早期形态发生过程中正确关闭视神经裂缝所必需的[15]。而转录因子c-Jun是JNK的主要目标,由于Jun是转录因子,所以Jun可能是控制了导致Bax基因激活的表达,一旦激活,Bax就会对线粒体造成无法修复的损害,同时会引发下游事件,从而结束一个垂死的神经节细胞[16]。Jun是RGCs的一个主要转录中心,控制着生存、支持死亡和支持再生的途径。因此,JNK/c-Jun信号通路作为一个凋亡信号通路在轴突损伤和神经元胞体变性之间有一个重要的作用[17]。最新研究表明,DDIT3(DNA damage-inducible transcript 3)和Jun具有相似功能,同样是RGC中独立调节的凋亡信号分子,并与Jun共同占据了轴突损伤后RGCs中绝大多数凋亡信号。与Jun或DDIT3单独缺陷相比,Jun和DDIT3联合缺失对RGCs的长期存活提供了更大的保护[18]。JNK/c-Jun信号通路的激活还可以在多种细胞应激下发生,例如在阿尔茨海默病(AD)患者的视网膜和脑中,Aβ寡聚体、P-APP和P-Tau特异性地积聚在RGCs层激活JNK途径,从而导致RGCs变性[19]。从神经损伤到神经营养缺失的DLK(dual leucine zipper kinase)激活,导致了c-Jun N-末端激酶(JNK)信号的激活[20];ONC通过TNF-α在RGC中诱导激活了JNK信号通路等。此外, 除了c-Jun,胞质内还有Bcl-2家族(Bcl-2、Bcl-XL、Bim、BAD),核内有ATF-2、Elk-1、c-My2 等蛋白亦为其底物[21]。
2.2钙通道抑制与JNK/c-Jun信号通路活性的关系根据目前的研究指出,钙离子流入是激活JNK/c-Jun信号通路的一种附加机制。钙离子信号在MAPK磷酸酶(MKP)mRNA的转录延伸中起着重要作用[22],并且已知其可以上调MKP-1[23]。轴突损伤导致钙离子增加,钙离子增加引发了MAPKs的激活,并显著激活JNK信号通路[24]。已有研究表明,JNK信号传导在损伤的轴突变性早期发挥作用[25]。在其早期激活之后,以Jun磷酸化的形式在轴突受损的RGC中观察到持续的JNK信号传导,在其磷酸化后立即损伤轴突。最近Ribas等[14]在轴突损伤模型应用钙通道抑制剂发现JNK/c-Jun信号通路的活性可以通过抑制钙通道而减弱。因此,钙通道抑制引起的JNK/c-Jun活性降低可能是减少轴突变性和RGC死亡的另一种机制。此外,KCa3.1,即中电导钙激活钾通道,参与许多细胞类型的激活。KCa3.1的激活在细胞活化过程中调节钾流出,膜超极化和钙流入[26]。之前的研究已经证明了KCa3.1在通过c-Jun/JNK MAPK信号通路来调节反应性星形胶质细胞的过程中起着关键作用[27]。KCa3.1通道的阻断也显示出减弱了Aβ诱导的JNK MAPK途径,导致阿尔茨海默病小鼠模型中脑部炎性因子IL-1β、TNF-α、iNOS和COX-2的下调[28]。这也从侧面反映出了钙通道对JNK信号通路的影响作用。
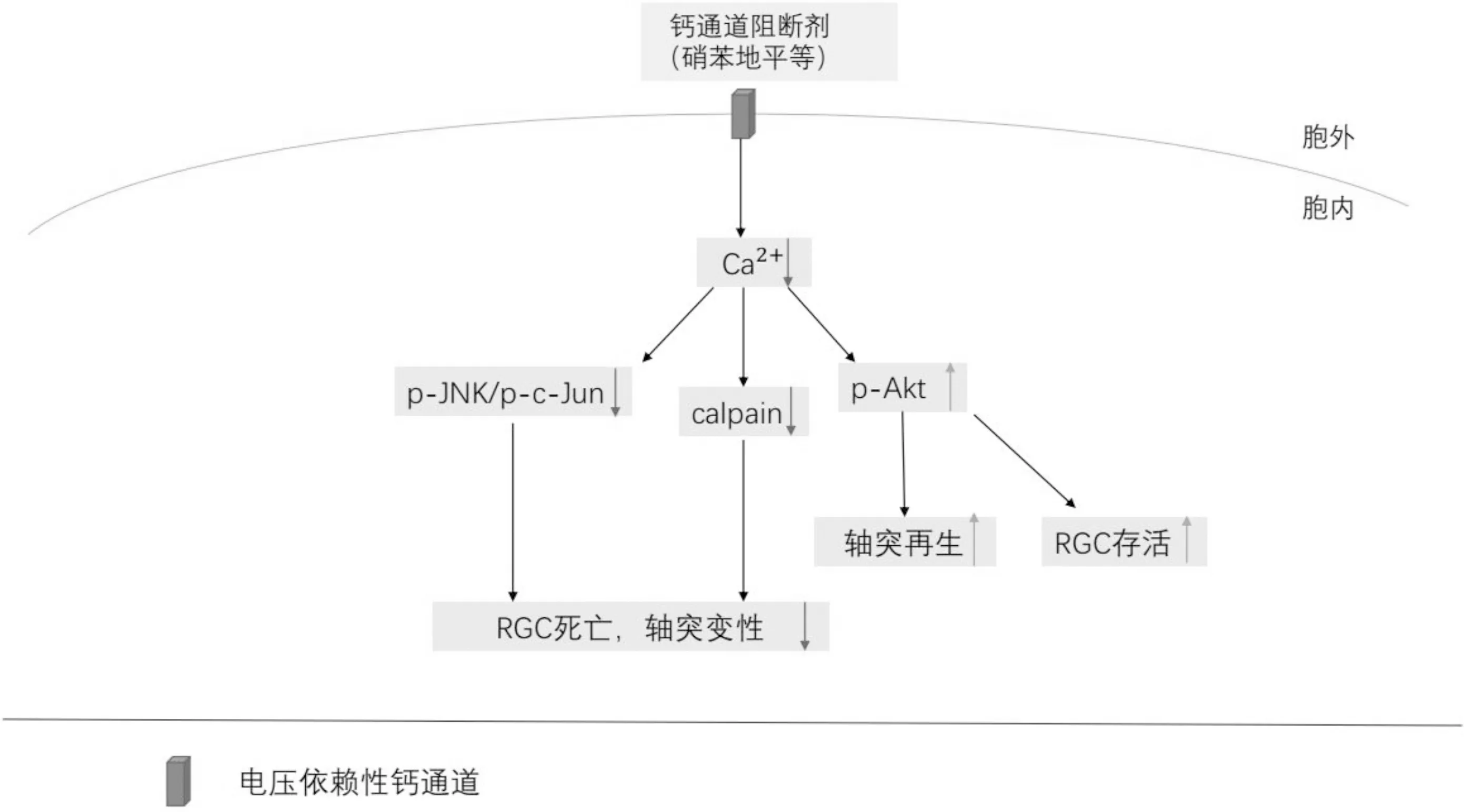
图1在钙通道阻断剂的作用下,胞内钙离子浓度下降,使钙蛋白酶和JNK/c-Jun活化减少,而Akt活化增加来减少轴突变性和RGC凋亡,促进轴突再生和RGC存活。
3 Akt信号途径
3.1 Akt信号通路的作用Akt,即蛋白激酶B(PKB),是一种丝氨酸/苏氨酸特异性蛋白激酶。Akt的激活由生长因子受体酪氨酸激酶刺激的磷脂酰肌醇3-激酶(PI3K)介导。Akt(PKB)是PI3K的一个下游效应因子。免疫荧光显示在DRG(dorsal root ganglia)细胞体和轴突生长锥中存在活化的Akt,PI3K抑制剂显著降低自发生长的神经元存活和轴突生长,神经生长因子或神经胶质细胞系衍生因子引起的生长增加也显着降低[29]。Akt在激活过程中瞬时定位于质膜,一旦激活,它就会诱导一些细胞核和胞质蛋白的磷酸化,从而调控多种细胞功能,包括细胞生长、存活、增殖和分化。Akt通过对Bcl-2家族成员和线粒体活性的调节发挥作用[30]。Akt可以加速轴突再生,促进轴突延伸和分支,抑制神经元凋亡[31]。目前研究认为,Akt主要通过促进哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、GSK-3等下游底物磷酸化而发挥生物学效应,其中包括抗凋亡、促细胞生存等[32-33]。在轴突横断的RGCs中激活mTOR信号传导促进轴突生长超过几毫米,一些轴突到达视交叉,即视觉通路的中点[34]。值得注意的是,磷酸酶和张力蛋白同系物PTEN(phosphatase and tensin homolog)在RGCs中的缺失促进了视神经损伤后轴突的再生[35]。RGCs中PTEN/mTORC1通路的调节促进视神经损伤后的轴突再生和RGC存活。糖原合成酶激酶3(GSK3)是Akt的信号传感器。Akt通过将GSK3α的Ser21磷酸化或GSK3β的Ser9磷酸化使GSK3的激酶活性失活。研究显示GSK3β是RGC中PTEN/Akt信号传导的主要下游靶标,而GSK3β的底物eIF2Be(真核翻译起始因子2Bε亚基)在所有真核细胞中翻译起始蛋白质,AKT-GSK3b-eIF2Be信号模块在确定成熟CNS神经元的内在轴突生长能力方面发挥着核心作用[36]。
3.2钙通道与Akt信号通路活性的关系p-Akt是一种支持生存的激酶[37],科研人员在大鼠视神经挤压伤(ONC)模型上研究了钙通道与Akt活性的关系,发现在钙通道抑制剂治疗的神经节细胞层中,可以观察到支持生存的Akt信号的激活(磷酸化),表明Akt在神经细胞的生存信号传递中有一个关键作用,以及包括RGC生存在内的轴突生长。因此,钙通道抑制引起的Akt活化的增加可以解释对RGC存活和轴突再生的影响[14]。Akt信号可以阻断轴突损伤后的不同退化信号,通过限制下游的JNK信号激活来调节退化的应答[3]。越来越多的证据表明,在受伤的视网膜中Akt信号通路在支持神经元存活方面有着重要的作用[38](图1)。
4总结与展望
钙离子的流入是轴突变性退化调节至关重要的因素。损伤后轴突中钙水平的升高代表了一种亚稳态的中间状态,而不是一个紧急且不可改变的毁灭过程。轴突的损失可以通过阻止钙的流入或钙的产生来抑制,即使不在早期也可发挥作用[39]。钙通道抑制剂可以刺激神经细胞内的钙离子泵,使胞内的钙离子浓度趋于稳定,减轻钙离子浓度异常对细胞的损伤[40]。目前大量研究表明,钙通道抑制剂的应用阻止钙在早期的升高,阻止了大部分轴突的退化变性,同时也有效减少细胞水肿和血管痉挛,避免视神经损伤的加重[41],所以通过阻断钙通道从而控制胞内钙的流入是非常重要的。总之,通过钙通道抑制对视神经损伤后轴突和RGCs进行保护是一种有效的手段,也是近年来全球学者研究的热门领域。当然,钙通道抑制剂对于损伤部位远端的继发变性效果不明显等一些缺点目前还没有攻克,对其机制的研究和开发还不够深入,临床试验和应用也还有待完善,但其对于以保持损伤后轴突的完整性为目标的干预是非常重要的。当下也是视神经损伤后针对轴突变性和RGC凋亡进行组合治疗策略的重要步骤。相信在不久的将来,在国内外学者的共同努力下,钙通道手段治疗ONC会逐步从科学理论走向临床实践,为视神经损伤的患者带来福音。
