应用代谢网络模型解析工业微生物胞内代谢
2019-10-31叶超徐楠陈修来刘立明
叶超,徐楠,陈修来,刘立明
应用代谢网络模型解析工业微生物胞内代谢
叶超1,2,徐楠3,陈修来1,2,刘立明1,2
1 江南大学 食品科学与技术国家重点实验室,江苏 无锡 214122 2 江南大学 工业生物技术教育部重点实验室,江苏 无锡 214122 3 扬州大学 生物科学与技术学院,江苏 扬州 225009
为了快速、高效地理解工业微生物胞内代谢特征,寻找潜在的代谢工程改造靶点,基因组规模代谢网络模型(GSMM) 作为一种系统生物学工具越来越受到人们的关注。文中在回顾GSMM 20年发展历程的基础上,分析了当前GSMM的研究现状,总结了GSMM的构建及分析方法,从预测细胞表型和指导代谢工程两个方面阐述了GSMM在解析工业微生物胞内代谢中的应用,并展望了GSMM未来的发展趋势。
微生物胞内代谢,生产性能,基因组规模代谢网络模型,系统生物学
基因组规模代谢网络模型 (Genome-scale metabolic model, GSMM) 作为一种数学模型,其本质是用于表征基因-蛋白-反应 (GPR) 三者之间的关系。GSMM包括一系列化学计量平衡的生化反应,通过形成一个矩阵S,将其转化成一个数学模型。矩阵S的每行代表代谢物,每列代表反应。GSMM已经广泛应用在分析网络特性、预测细胞表型、指导菌株设计、驱动模型发现、研究进化过程和分析相互作用等6个方面(图1)[1-2]。
工业微生物胞内代谢活动具有较高的复杂性,单一的研究手段难以系统地理解其调控机制,且无法高效地获取所需表型。目前利用GSMM分析工业微生物胞内的代谢调控,尤其是筛选代谢工程改造靶点,提高目标产物产量方面已经取得了一定的进展。例如,Nocon等利用MOMA算法鉴定出乙醇脱氢酶是毕赤酵母中过量生成胞质人超氧化物歧化酶(hSOD) 基因敲除靶点,实验证实的敲除使得hSOD产量增加了20%[3]。Song等利用流量响应分析方法,筛选出扩增磷酸烯醇丙酮酸羧化酶() 使富马酸产量提高了2.8倍[4]。这些基于GSMM预测和实验结合的方法,在乙醇[5-6]、丁醇[7-8]、琥珀酸[9-10]、乳酸[11]、番茄红素[12]、氨基酸[13-14]、香兰素[15]和1,4-丁二醇[16]等产品生产中展现了巨大的应用前景。
本文综述了目前GSMM的研究进展,比较了目前常见的模型构建及评价标准,总结出流量平衡类和遗传扰动类模型分析方法的应用范围。进一步结合这些模型分析工具,系统地阐述GSMM在工业微生物表型预测、代谢改造等胞内代谢研究中的应用。同时展望了GSMM的未来发展趋势,以期能够利用系统生物学工具,为“湿实验”提供理论指导。
1 代谢网络模型的发展
1.1 代谢网络模型的研究现状
自1999年完成流感嗜血杆菌GSMM的构建以来,GSMM经历了20年的发展,截至2019年3月,已经有超过153种微生物,348个GSMMs完成了构建(图2)[1-17]。其中一些模式菌株的GSMMs,如大肠杆菌K12和酿酒酵母S288c,经过不断的修正和完善,分别构建了6个和12个GSMMs[17-18]。改进后的GSMMs,不仅增加了模型规模(基因、反应、代谢物),而且提高了模型预测结果的准确性。如最新.模型ML1515预测基因敲除表型的准确率为93.4%,相较JO1366准确率89.8%提高了3.6%[19]。
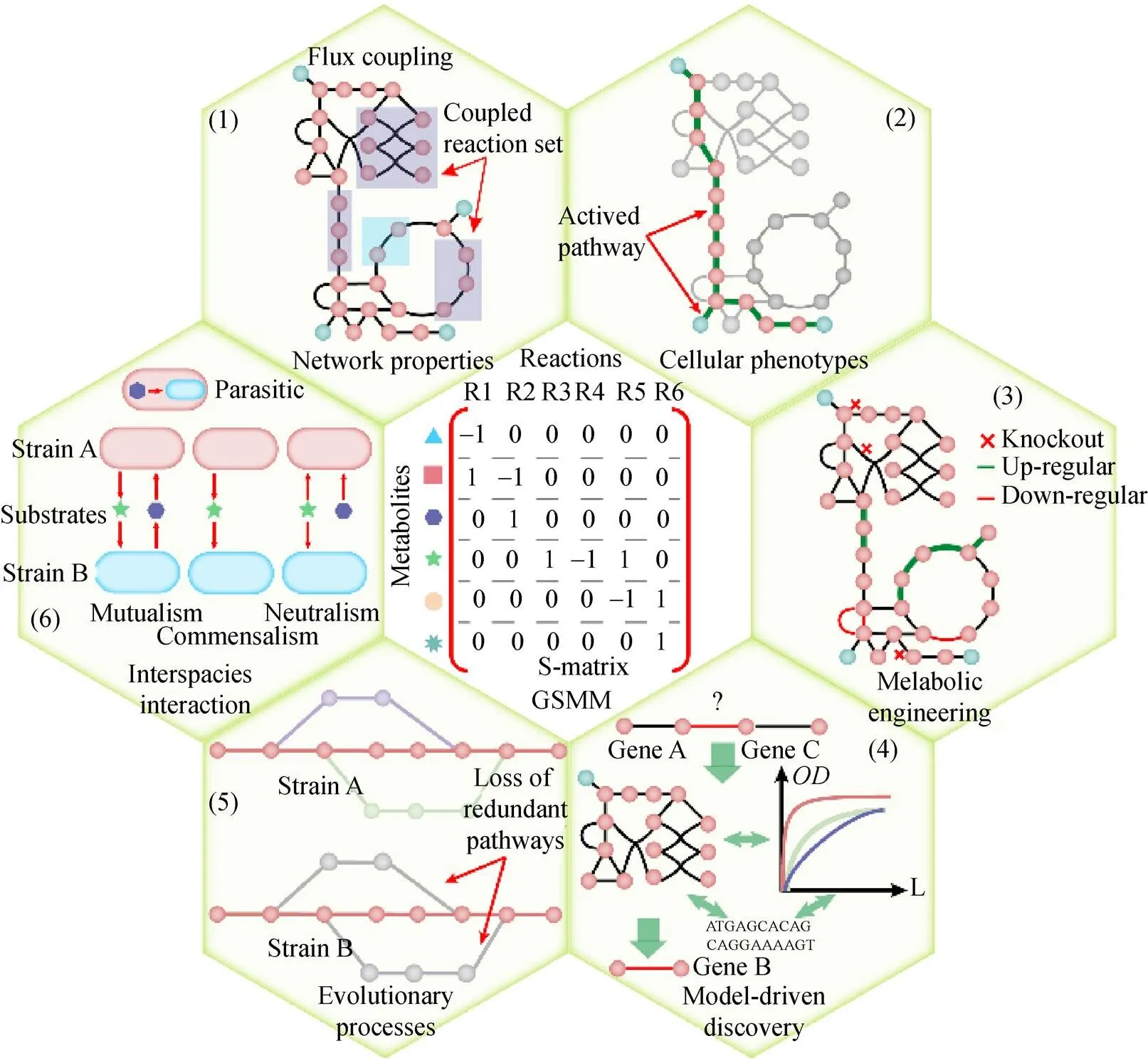
图1 GSMM在工业微生物中的应用
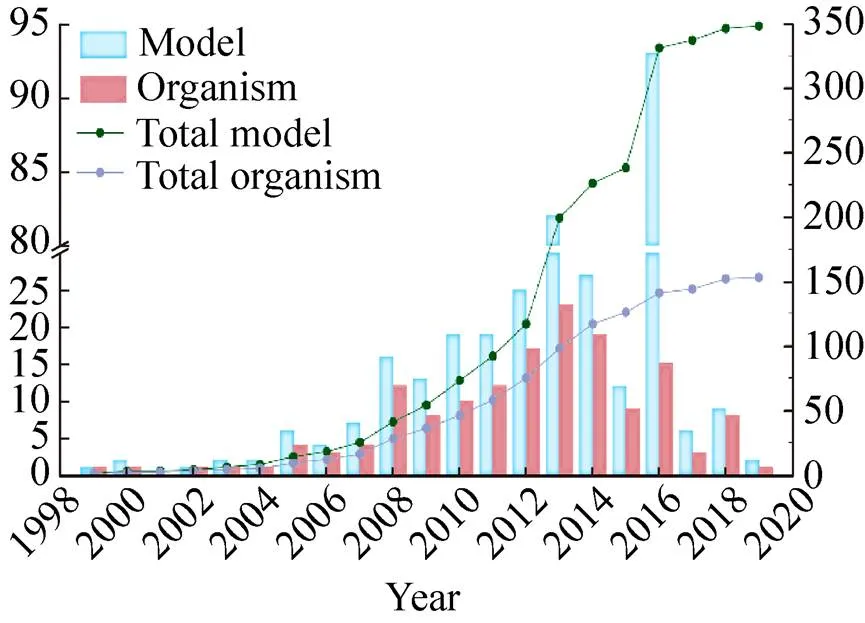
图2 已发表代谢网络模型的统计
由于传统的GSMM主要是通过对底物的利用作为约束条件进行模拟分析,鲁棒性分析碳源吸收速率与细胞生长之间的关系通常呈线性相关。表明随着碳源吸收速率的增加,细胞比生长速率可以一直增加,最终远大于菌株的最大比生长速率。因此,为了提高模型预测结果的准确性,需要在传统GSMM的基础上整合其他方法。目前的改进方法主要有4个方面:1) 整合组学数据[20-22]:如通过整合转录组学数据,得到基因表达模型(ME-model),预测结果的准确性从91.2%提高至92.3%[20];2) 添加各种约束条件[23-26]:将动力学参数整合至.核心GSMM中,模拟结果与实验数据的皮尔森相关系数达到了0.84[23];3) 整合多种生物模型:在GSMM基础上整合基因表达数据、转录调控模型、信号转导网络,得到.整合模型,皮尔森相关系数提高了2倍[27];4) 构建微生物全细胞模型:利用生殖支原体全细胞模型不仅可以从单细胞水平描述一个细胞周期内个体分子及其相互作用,而且实现了一系列可观察到的细胞行为的准确预测[28]。
1.2 代谢网络模型的发展趋势
1.2.1 互作网络模型
自然界中的微生物相互作用关系主要存在互生、共生、竞争和寄生。通过构建微生物群体互作模型,模拟不同物种之间的代谢物交换,已经被用于鉴定微生物之间的相互作用关系。通过构建维生素C混菌发酵模型WZ-KV-663-BM-1055,共培养条件下,生酮基古龙酸菌和巨大芽孢杆菌的最大比生长速率分别比单独培养提高了1.5倍和6.6倍,表明两菌之间存在共生关系,并且是通过代谢物交换实现的:.能为.提供6种氨基酸、3种核苷酸、6种维生素和辅因子、3种有机酸以及甘油等营养物质;而.通过分泌苯丙氨酸、富马酸和甲酸促进.生长[29]。类似地,通过分别构建木质纤维素生产丁醇菌株丙酮丁醇梭菌和解纤维梭菌的GSMMs,模拟共培养条件下丁醇发酵过程,发现.和.之间也是代谢物交换而实现共生(图3)[30]。
1.2.2 泛基因组模型
泛基因组(Pan-genome) 是指某一物种全部基因的总称,包括核心基因组(Core genome) 和附加基因组(Dispensable genome)。通过构建泛基因组模型,对核心基因和附加基因进行组合分析,比较同一物种不同菌株之间的表型差异,从而分析菌株特异性。目前已经完成构建的泛基因组模型主要有:包含了55株.的泛基因组模型[31]、64株金黄色葡萄球菌泛基因组模型[32]以及410株沙门氏菌属[33]的泛基因组模型。由于涉及的菌株都是致病性菌株,主要是利用泛基因组模型预测不同菌株之间的代谢能力差异,从而鉴定致病性。例如,在泛基因组模型中,通过预测410株在530种环境中的生长能力,鉴定出这些存在显著差异的代谢途径包括碳代谢和细胞壁合成,并且代谢特异性与每个菌株的血清型和分离宿主相对应[33]。
1.2.3 宏基因组模型
宏基因组 (Meta-genome) 是指特定环境中全部微生物的总DNA。物种丰富度(用来描述和量化微生物群落,反映特定区域物种的数量),是宏基因组数据的重要评价指标。在单个微生物GSMM的基础上,通过构建宏基因组网络模型,一方面可以结合模型分析菌群在不同环境中的代谢特征,另一方面可以预测不同微生物之间的相互作用关系。目前已经构建完成的宏基因组模型是包含773个肠道微生物的宏基因组模型[34]和包含1 562个人类相关微生物的宏基因组模型[35]。考虑到宏基因组数据的物种丰富度,需要批量构建成百上千个GSMMs,因此在构建宏基因组模型的过程中需要开发特定的方法,如AGORA[34]和MOMBO[35]。这些算法的特点在于实现模型批量构建的过程中,建立了特定的模型质量评价体系,从而满足模拟分析。
2 代谢网络模型的分析
2.1 模型构建
根据Palsson实验室发表在上的GSMM构建教程[36-37],GSMM的构建主要包括4个部分:粗模型的构建、模型的精炼、模型的数学转换、模型的调试和验证,涉及102个具体步骤。而GSMM的构建方式有3种:手动构建、自动构建和半自动构建。手动构建方法依赖于基因组注释结果,结合KEGG中的pathway map,对每个代谢途径涉及的基因-蛋白-反应进行收集与整理,这种方法的优势在于模型的准确性较高,缺点则是模型构建过程耗时。自动构建方法则是通过已有的工具,提交相关的信息,如基因组序列,从而自动得到目的菌株的GSMM,目前已经开发了许多工具,如COBRA[37]、RAVEN[38]、Model SEED[39]和IMGMD[17],用于GSMM的自动化构建。这种方式的优势在于可以实现模型的快速、批量构建,缺点则是构建得到的模型准确性低。半自动构建的方法则是先通过一些自动化构建工具得到GSMM,然后在该模型的基础上进行手动精炼,得到精确的模型。
在完成模型的构建后,需要进行一系列的验证,从而判断其覆盖范围是否完整,以及与现有的模型相比是否具有更好的预测能力。常见的模型评价指标主要有:通用指标、连通度指标、生长指标和基因敲除指标 (表1)。前两个属于描述模型的指标 (定量和定性),后两个是预测生物行为的指标[40]。通过将不同培养条件下的模拟结果与实验值进行比较,从而判断模型模拟结果的准确性。
2.2 模型分析
2.2.1 流量平衡分析类算法
代谢流量(Flux) 是胞内分子通过代谢途径的转换率,受到代谢途径中涉及的酶调控。流量平衡分析(FBA) 作为最基本的数学算法,目前已经被广泛应用于计算最优条件下的生长速率或者代谢产物合成速率[41]。然而由于FBA算法局限性是只能模拟稳态条件下的流量分布,并且通过底物的吸收速率作为约束条件,限制了FBA预测流量分布的准确性。因此,在FBA的基础上开发了一系列的算法对代谢流量进行额外的约束(表2)。这些算法一方面提高了模型预测流量分布结果的准确性,例如,通过整合转录调控信息,开发了rFBA算法,根据培养基中的代谢物浓度对反应流量添加布尔约束,能够解释、分析和预测转录调控在系统水平上对细胞代谢的影响[42]。在rFBA的基础上,进一步整合常微分方程(ODEs),开发了iFBA算法[43],模拟结果表明,相较于rFBA,iFBA对334个单基因扰动和野生型的预测更准确。另一方面拓展了模型的应用,如dFBA算法将FBA扩展至能够考虑动力学因素,实现代谢流量的动态模拟。应用该算法,准确预测了.在葡萄糖分批补料条件下的二次生长情况[44]。cFBA则通过将反应化学计量数、热力学和生态系统作为约束条件,用于研究微生物群体的代谢行为[45]。

表1 用于评价代谢网络模型的主要指标

表2 常见的流量平衡分析方法
2.2.2 遗传扰动类算法
微生物遗传改造主要是通过对微生物基因组进行修饰,改变基因型,从而获得不同表型微生物。结合GSMM,开发相应的算法可以实现对这些扰动的模拟(表3)。这些算法可以分为基因敲除、添加、上调/下调或者异源途径导入等。Optknock是典型的基因敲除方法,是基于双目标方程,即分别对生物量方程和目标产物求解,筛选出能够提高目标产物的基因敲除靶点[51]。在OptKnock的基础上进行改进,得到了ReacKnock 算法,能够实现多达20个基因敲除靶点的筛选[52]。OptSwap作为基因添加类唯一的算法,主要是一种确定氧化还原酶辅因子特异性(NAD(H)和NADP(H)) 的最佳修饰的方法,用于微生物生产菌株的设计[53]。在基因上调、下调方面,OptForce通过比较突变菌株和野生型的反应流量差异,从而确定上调或下调的靶点,实现目标产物的过量合成[54]。而k-OptForce则是OptForce的扩展,通过整合酶动力学常数,预测酶编码基因的上调或下调[55]。异源表达相关的算法目前有OptStrain和SimOptStrain。前者通过在包含一系列酶催化反应的反应库中检索,筛选能够满足最大产物合成速率下的最小修饰路径添加至宿主细胞中[56]。后者则是不仅能够实现异源途径的添加,同时鉴定出宿主细胞中需要删除的反应[57]。

表3 常见的模拟菌株设计方法
3 代谢网络模型在解析胞内代谢中的应用
3.1 预测细胞表型
在工业菌株生产过程中,微生物的表型是基因型和外部环境共同作用的结果。微生物的表型主要涉及细胞生长、能量利用、底物利用、产物合成和基因必需性等方面。利用GSMM对细胞表型进行预测,从而理解微生物的表型潜力。
为了预测微生物的细胞生长,利用FBA算法,计算稳态条件下的各个反应的流量分布。通过设定不同底物的吸收速率,以生物量方程为目标方程求解,从而计算出.的最大比生长速率()[41]。
通过改变目标方程,探讨微生物代谢在不同目标方程下的流量分布,以检验满足细胞功能背后的驱动力,进而理解能量利用与细胞次优行为之间的关系。通过评价11种目标方程下的流量分布,模拟不同条件下的最大ATP得率,与6种环境下的C13标记实验进行比较,证实模型能够准确预测微生物胞内能量利用[67]。
通过改变培养条件,模拟不同培养基中底物利用对微生物生长的影响,从而鉴定最优培养基。利用维生素C生产菌株.WSH001模型WZ663,通过FBA模拟,发现从完全培养基中分别去除甘氨酸、半胱氨酸、甲硫氨酸、色氨酸、腺嘌呤、胸腺嘧啶、硫胺素和泛酸会导致细胞生长分别下降1%、21%、16%、1%、26%、57%、73%和24%。基于这些结果开发了.最小合成培养基,不仅满足了.的生长,而且2-酮基-L-古龙酸产量达到了完全培养基条件下的96.5%[68]。
利用鲁棒性分析算法,对生物量方程进行约束,以产物合成反应作为目标方程求解,能够得到一个解空间,反映了不同生长速率下的产物合成能力。结合OptKnock算法,筛选.过量生成乳酸的靶点,结合适应性进化策略,最终得到能够在M9培养基中同时满足快速生长和乳酸大量分泌的.菌株[11]。
利用单基因敲除程序,对微生物的必需基因鉴定。通过比较不同培养条件下的必需基因差异,从而理解基因的绝对必需性和相对必需性。在最小培养基中,高山被孢霉的模型CY1106中有86个基因被鉴定为必需基因;而在酵母提取物培养基中,只有49个基因是必需基因。进一步分析,前者有36.05%的必需基因属于氨基酸代谢途径,而后者有23.26%的必需基因参与核苷酸代谢[69]。表明在营养丰富的培养条件下,氨基酸可以通过从培养基中直接摄入,不需要从头合成,因此氨基酸代谢相关的基因是相对必需的。
综上所述,在细胞表型预测方面,GSMM展现了巨大的应用前景。利用GSMM不仅可以模拟微生物在不同培养条件下的生长情况,为发酵过程优化提供参考,而且实现了胞内代谢流分布的定量分析,为代谢瓶颈的鉴定提供了理论依据。
3.2 指导代谢工程
代谢工程改造目标菌株,提高产量是一种有效策略,已经被广泛地应用[70]。但是由于缺乏对微生物整个代谢活动的系统认识,单一水平上的基因改造策略难以高效地实现目标。利用GSMM,结合不同的遗传改造方法,可以从全局水平模拟:1) 竞争路径的消除;2) 合成路径关键基因的强化;3) 反馈抑制的消除;4) 外源路径的导入;5) 辅因子优化等改造策略对目标产物的影响,为系统代谢工程提供指导[71]。
为了消除竞争路径,通过一系列的算法筛选基因敲除靶点,对基因敲除结果的组合优化,从而提高目标产品的产量。利用OptCouple,鉴定出大肠杆菌分别过量生产丙酸、衣康酸的一系列基因敲除靶点,这些靶点大多存在于其竞争途径,此外还鉴定出甲基化修饰提高产量的靶点[61]。
在基因敲除菌株的基础上,表达特定基因,模拟合成路径关键基因的强化,分析基因表达对产物合成的影响。利用OptORF算法鉴定出敲除转录因子,同时敲除基因、和,在此基础上过量表达,使得乙醇得率从39.3%提高至86.2%[63]。
反馈抑制的消除主要是通过不同的算法模拟代谢路径中某个基因的上调或下调,从而使碳流更多地流向目标产物。利用OptForce算法,对不同链长脂肪酸合成过程中的上调、下调、敲除靶点进行了鉴定。发现上调和acyl-ACP硫酯酶,同时敲除,最小M9培养基中C14-16脂肪酸的产量达到了1.7 g/L,得率为0.14 g/g葡萄糖(最大理论得率的39%)[72]。同样地,利用该方法鉴定出敲除和,同时上调ACC、PGK、GAPD和PDH的表达,能够使胞内丙酰辅酶A的含量提高3.1倍[73]。
对于非天然产物的合成,通常涉及异源途径的导入。利用SimOptStrain鉴定出在.中敲除、和的同时,引入氧代戊二酸脱氢酶(EC: 1.2.1.52) 催化的反应(2-酮戊二酸+ CoA+NADP+=琥珀酰辅酶A+CO2+NADPH),使得琥珀酸得率达到了最大理论得率的32.5%[57]。
通过改善产物合成路径中关键酶的辅因子的偏好性,消除辅因子失衡所造成的负面效应,有助于构建高效的产物合成路径。基于OptSwap算法设计和优化光滑球拟酵母生产丙酮酸,得到了12种辅因子交换和敲除策略,导致丙酮酸合成速率从0最高增加至 20.42 mmol/(g DW·h)[74]。
综上所述,基于GSMM,筛选不同代谢改造靶点并组合,用于指导代谢工程,进而通过实验进行验证。应用这种策略,已经成功地应用在有机酸、氨基酸、醇类等化学品的生产中。模型与实验相结合,不仅实现了菌株改造的理性设计,提高了代谢工程改造效率,而且能够从全局水平考虑遗传扰动对整个微生物胞内代谢的影响,有助于实现代谢流的精准调控。
4 结论与展望
针对工业微生物胞内代谢的研究,国内外研究人员采用生化工程和代谢工程等策略,开展了卓有成效的研究。结合GSMM,可以将理性设计与实验操作结合,从而高效地获得工业生产菌株。GSMM经过20年的发展,一方面通过不断添加约束条件,提高了模型预测结果的准确性,另一方面从单一菌株的模型发展至宏基因组模型。随着一系列模型构建工具和分析方法的开发,不仅实现了微生物GSMM的快速构建,而且能够从预测细胞表型、指导代谢工程这两个方面对微生物胞内代谢活动进行系统分析。
随着GSMM构建工具的不断开发和完善,目前已经可以实现GSMM的快速、批量构建,但是受限于模型构建方法及构建团队自身水平的限制,导致构建的GSMMs质量参差不齐,此外,这些GSMMs中的反应和代谢物列表中的展现形式也缺乏统一,限制了GSMM的进一步应用。虽然目前已有一些数据库,如BIGG Models实现了已发表GSMMs的标准化,但是该数据库目前仅收录了85个模型[18]。因此亟需建立相应的标准对模型进行统一,并且搭建功能更全面的数据库平台,提供GSMMs的检索和下载。
虽然GSMM已经被广泛地应用在多个方面,但是这些模型的约束参数都是固定的。而最新发展的机器学习(Machine learning) 方法,基于黑箱理论(Black-box),不需要对约束参数进行设定,只需要提供一系列的训练集(Training data),就能够对参数进行自动修正,从而实现模型的精确预测。此外,为了克服黑箱机器学习方法无法直接揭示生物分子与细胞表型之间的相互作用关系的不足,Yang等进一步开发了白箱理论(White-box) 用于揭示抗生素作用机制[75]。机器学习方法,作为一种新兴的技术,在现代社会展现了巨大的应用潜力,已经广泛地应用在语言识别、视觉对象识别、物体检测、药物发现和基因组分析等方面[76-78]。因此,采用机器学习方法对GSMM进行分析,除了进一步实现代谢流调控的精准预测,还可以用于复杂环境下相互作用关系的表征,则是未来GSMM的发展趋势。
[1] Kim WJ, Kim HU, Lee SY. Current state and applications of microbial genome-scale metabolic models. Curr Opin Syst Biol, 2017, 2: 10–18.
[2] O'Brien EJ, Monk JM, Palsson BO. Using genome-scale models to predict biological capabilities. Cell, 2015, 161(5): 971–987.
[3] Nocon J, Steiger MG, Pfeffer M, et al. Model based engineering ofcentral metabolism enhances recombinant protein production. Metab Eng, 2014, 24: 129–138.
[4] Song CW, Kim DI, Choi S, et al. Metabolic engineering offor the production of fumaric acid. Biotechnol Bioeng, 2013, 110(7): 2025–2034.
[5] Dikicioglu D, Pir P, Onsan ZI, et al. Integration of metabolic modeling and phenotypic data in evaluation and improvement of ethanol production using respiration-deficient mutants of. Appl Environ Microbiol, 2008, 74(18): 5809–5816.
[6] Lee KY, Park JM, Kim TY, et al. The genome-scale metabolic network analysis ofZM4 explains physiological features and suggests ethanol and succinic acid production strategies. Microb Cell Fact, 2010, 9: 94.
[7] Lee J, Yun H, Feist AM, et al. Genome-scale reconstruction andanalysis of theATCC 824 metabolic network. Appl Microbiol Biotechnol, 2008, 80(5): 849–862.
[8] Ranganathan S, Maranas CD. Microbial 1-butanol production: identification of non-native production routes andengineering interventions. Biotechnol J, 2010, 5(7): 716–725.
[9] Lee SJ, Lee DY, Kim TY, et al. Metabolic engineering offor enhanced production of succinic acid, based on genome comparison andgene knockout simulation. Appl Environ Microbiol, 2005, 71(12): 7880–7887.
[10] Mienda BS, Shamsir MS, Illias RM. Model-guided metabolic gene knockout of gnd for enhanced succinate production infrom glucose and glycerol substrates. Comput Biol Chem, 2016, 61: 130–137.
[11] Fong SS, Burgard AP, Herring CD, et al.design and adaptive evolution offor production of lactic acid. Biotechnol Bioeng, 2005, 91(5): 643–648.
[12] Choi HS, Lee SY, Kim TY, et al.identification of gene amplification targets for improvement of lycopene production. Appl Environ Microbiol, 2010, 76(10): 3097–3105.
[13] Lee KH, Park JH, Kim TY, et al. Systems metabolic engineering offorL-threonine production. Mol Syst Biol, 2007, 3(1): 149.
[14] Park JH, Lee KH, Kim TY, et al. Metabolic engineering offor the production ofL-valine based on transcriptome analysis andgene knockout simulation. Proc Natl Acad Sci USA, 2007, 104(19): 7797–7802.
[15] Brochado AR, Matos C, Møller BL, et al. Improved vanillin production in baker’s yeast throughdesign. Microb Cell Fact, 2010, 9: 84.
[16] Yim H, Haselbeck R, Niu W, et al. Metabolic engineering offor direct production of 1,4-butanediol. Nat Chem Biol, 2011, 7(7): 445–452.
[17] Ye C, Xu N, Dong C, et al. IMGMD: a platform for the integration and standardisation ofmicrobial genome-scale metabolic models. Sci Rep, 2017, 7(1): 727.
[18] King ZA, Lu J, Dräger A, et al. BiGG Models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res, 2016, 44(D1): D515–D522.
[19] Monk JM, Lloyd CJ, Brunk E, et al.ML1515, a knowledgebase that computestraits. Nat Biotechnol, 2017, 35(10): 904–908.
[20] O’Brien EJ, Lerman JA, Chang RL, et al. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol Syst Biol, 2013, 9(1): 693.
[21] Yizhak K, Benyamini T, Liebermeister W, et al. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics, 2010, 26(12): i255–i260.
[22] Feng XY, Zhao HM. Investigating xylose metabolism in recombinantvia13C metabolic flux analysis. Microb Cell Fact, 2013, 12: 114.
[23] Khodayari A, Maranas CD. A genome-scalekinetic metabolic model k-ecoli457 satisfying flux data for multiple mutant strains. Nat Commun, 2016, 7: 13806.
[24] Krumholz EW, Libourel IGL. Thermodynamic constraints improve metabolic networks. Biophys J, 2017, 113(3): 679–689.
[25] Sánchez BJ, Zhang C, Nilsson A, et al. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol Syst Biol, 2017, 13(8): 935.
[26] Brunk E, Sahoo S, Zielinski DC, et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat Biotechnol, 2018, 36(3): 272–281.
[27] Carrera J, Estrela R, Luo J, et al. An integrative, multi-scale, genome-wide model reveals the phenotypic landscape of. Mol Syst Biol, 2014, 10(7): 735.
[28] Karr JR, Sanghvi JC, Macklin DN, et al. A whole-cell computational model predicts phenotype from genotype. Cell, 2012, 150(2): 389–401.
[29] Ye C, Zou W, Xu N, et al. Metabolic model reconstruction and analysis of an artificial microbial ecosystem for vitamin C production. J Biotechnol, 2014, 182–183: 61–67.
[30] Salimi F, Zhuang K, Mahadevan R. Genome-scale metabolic modeling of a clostridial co-culture for consolidated bioprocessing. Biotechnol J, 2010, 5(7): 726–738.
[31] Monk JM, Charusanti P, Aziz RK, et al. Genome-scale metabolic reconstructions of multiplestrains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci USA, 2013, 110(50): 20338–20343.
[32] Bosi E, Monk JM, Aziz RK, et al. Comparative genome-scale modelling ofstrains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci USA, 2016, 113(26): E3801–E3809.
[33] Seif Y, Kavvas E, Lachance JC, et al. Genome-scale metabolic reconstructions of multiplestrains reveal serovar-specific metabolic traits. Nat Commun, 2018, 9(1): 3771.
[34] Magnúsdóttir S, Heinken A, Kutt L, et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol, 2017, 35(1): 81–89.
[35] Garza DR, Van Verk MC, Huynen MA, et al. Towards predicting the environmental metabolome from metagenomics with a mechanistic model. Nat Microbiol, 2018, 3(4): 456–460.
[36] Schellenberger J, Que R, Fleming RMT, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc, 2011, 6(9): 1290–1307.
[37] Heirendt L, Arreckx S, Pfau T, et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat Protoc, 2019, 14(3): 639–702.
[38] Wang H, Marcišauskas S, Sánchez BJ, et al. RAVEN 2.0: a versatile toolbox for metabolic network reconstruction and a case study on. PLoS Comput Biol, 2018, 14(10): e1006541.
[39] Alper HS. Systems Metabolic Engineering. New York: Humana Press, 2013: 17–45.
[40] Sanchez BJ, Nielsen J. Genome scale models of yeast: towards standardized evaluation and consistent omic integration. Integr Biol (Camb), 2015, 7(8): 846–858.
[41] Orth JD, Thiele I, Palsson BØ. What is flux balance analysis? Nat Biotechnol, 2010, 28(3): 245–248.
[42] Covert MW, Schilling CH, Palsson B. Regulation of gene expression in flux balance models of metabolism. J Theor Biol, 2001, 213(1): 73–88.
[43] Covert MW, Xiao N, Chen TJ, et al. Integrating metabolic, transcriptional regulatory and signal transduction models in. Bioinformatics, 2008, 24(18): 2044–2050.
[44] Mahadevan R, Edwards JS, Doyle III FJ. Dynamic flux balance analysis of diauxic growth in. Biophys J, 2002, 83(3): 1331–1340.
[45] Khandelwal RA, Olivier BG, Roling WFM, et al. Community flux balance analysis for microbial consortia at balanced growth. PLoS ONE, 2013, 8(5): e64567.
[46] Lewis NE, Hixson KK, Conrad TM, et al. Omic data from evolved.are consistent with computed optimal growth from genome-scale models. Mol Syst Biol, 2010, 6(1): 390.
[47] Thiele I, Fleming RMT, Bordbar A, et al. Functional characterization of alternate optimal solutions of’s transcriptional and translational machinery. Biophys J, 2010, 98(10): 2072–2081.
[48] Benyamini T, Folger O, Ruppin E, et al. Flux balance analysis accounting for metabolite dilution. Genome Biol, 2010, 11(4): R43.
[49] Kelk SM, Olivier BG, Stougie L, et al. Optimal flux spaces of genome-scale stoichiometric models are determined by a few subnetworks. Sci Rep, 2012, 2: 580.
[50] Smallbone K, Simeonidis E. Flux balance analysis: a geometric perspective. J Theor Biol, 2009, 258(2): 311–315.
[51] Burgard AP, Pharkya P, Maranas CD. OptKnock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng, 2003, 84(6): 647–657.
[52] Xu ZX, Zheng P, Sun JB, et al. ReacKnock: identifying reaction deletion strategies for microbial strain optimization based on genome-scale metabolic network. PLoS ONE, 2013, 8(12): e72150.
[53] King ZA, Feist AM. Optimizing cofactor specificity of oxidoreductase enzymes for the generation of microbial production strains—OptSwap. Ind Biotechnol, 2013, 9(4): 236–246.
[54] Ranganathan S, Suthers PF, Maranas CD. OptForce: an optimization procedure for identifying all genetic manipulations leading to targeted overproductions. PLoS Comput Biol, 2010, 6(4): e1000744.
[55] Chowdhury A, Zomorrodi AR, Maranas CD. k-OptForce: integrating kinetics with flux balance analysis for strain design. PLoS Comput Biol, 2014, 10(2): e1003487.
[56] Pharkya P, Burgard AP, Maranas CD. OptStrain: a computational framework for redesign of microbial production systems. Genome Res, 2004, 14(11): 2367–2376.
[57] Kim J, Reed JL, Maravelias CT. Large-scale bi-level strain design approaches and mixed-integer programming solution techniques. PLoS ONE, 2011, 6(9): e24162.
[58] Segrè D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA, 2002, 99(23): 15112–15117.
[59] Ren SG, Zeng B, Qian XN. Adaptive bi-level programming for optimal gene knockouts for targeted overproduction under phenotypic constraints. BMC Bioinformatics, 2013, 14(S2): S17.
[60] Tepper N, Shlomi T. Predicting metabolic engineering knockout strategies for chemical production: accounting for competing pathways. Bioinformatics, 2010, 26(4): 536–543.
[61] Jensen K, Broeken V, Hansen ASL, et al. OptCouple: joint simulation of gene knockouts, insertions and medium modifications for prediction of growth-coupled strain designs. Metab Eng Commun, 2019, 8: e00087.
[62] Patil KR, Rocha I, Förster J, et al. Evolutionary programming as a platform formetabolic engineering. BMC Bioinformatics, 2005, 6: 308.
[63] Kim J, Reed JL. OptORF: optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst Biol, 2010, 4: 53.
[64] Gu DQ, Zhang C, Zhou SG, et al. IdealKnock: a framework for efficiently identifying knockout strategies leading to targeted overproduction. Comput Biol Chem, 2016, 61: 229–237.
[65] Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng, 2006, 8(1): 1–13.
[66] Rockwell G, Guido NJ, Church GM. Redirector: designing cell factories by reconstructing the metabolic objective. PLoS Comput Biol, 2013, 9(1): e1002882.
[67] Schuetz R, Kuepfer L, Sauer U. Systematic evaluation of objective functions for predicting intracellular fluxes in. Mol Syst Biol, 2007, 3(1): 119.
[68] Fan SC, Zhang ZY, Zou W, et al. Development of a minimal chemically defined medium forWSH001 based on its genome-scale metabolic model. J Biotechnol, 2014, 169: 15–22.
[69] Ye C, Xu N, Chen HQ, et al. Reconstruction and analysis of a genome-scale metabolic model of the oleaginous fungus. BMC Syst Biol, 2015, 9: 1.
[70] Chen XL, Gao C, Guo L, et al. DCEO Biotechnology: tools to design, construct, evaluate, and optimize the metabolic pathway for biosynthesis of chemicals. Chem Rev, 2018, 118(1): 4–72.
[71] Choi KR, Jang WD, Yang D, et al. Systems metabolic engineering strategies: integrating systems and synthetic biology with metabolic engineering. Trends Biotechnol, 2019, 37(8): 817–837.
[72] Ranganathan S, Tee TW, Chowdhury A, et al. An integrated computational and experimental study for overproducing fatty acids in. Metab Eng, 2012, 14(6): 687–704.
[73] Xu P, Ranganathan S, Fowler ZL, et al. Genome-scale metabolic network modeling results in minimal interventions that cooperatively force carbon flux towards malonyl-CoA. Metab Eng, 2011, 13(5): 578–587.
[74] Xu N, Ye C, Chen XL, et al. Genome-scale metabolic modelling common cofactors metabolism in microorganisms. J Biotechnol, 2017, 251: 1–13.
[75] Yang JH, Wright SN, Hamblin M, et al. A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell, 2019, 177(6): 1649–1661.e9.
[76] LeCun Y, Bengio Y, Hinton G. Deep learning. Nature, 2015, 521(7553): 436–444.
[77] Moen E, Bannon D, Kudo T, et al. Deep learning for cellular image analysis. Nat Methods, 2019, doi: 10.1038/s41592-019-0403-1.
[78] Gazestani VH, Lewis NE. From genotype to phenotype: augmenting deep learning with networks and systems biology. Curr Opin Syst Biol, 2019, 15: 68–73.
Application of metabolic network model to analyze intracellular metabolism of industrial microorganisms
Chao Ye1,2, Nan Xu3, Xiulai Chen1,2, and Liming Liu1,2
1 State Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi 214122, Jiangsu, China 2 Key Laboratory of Industrial Biotechnology, Ministry of Education, Jiangnan University, Wuxi 214122, Jiangsu, China 3 College of Bioscience and Biotechnology, Yangzhou University, Yangzhou 225009,Jiangsu, China
To quickly and efficiently understand the intracellular metabolic characteristics of industrial microorganisms, and to find potential metabolic engineering targets, genome-scale metabolic network models (GSMMs) as a systems biology tool, are attracting more and more attention. We review here the 20-year history of metabolic network model, analyze the research status and development of GSMMs, summarize the methods for model construction and analysis, and emphasize the applications of metabolic network model for analyzing intracellular metabolic activity of microorganisms from cellular phenotypes, and metabolic engineering. Furthermore, we indicate future development trend of metabolic network model.
intracellular metabolic activity of microorganisms, production performance, genome-scale metabolic model, systems biology
10.13345/j.cjb.190257
刘立明 博士、教授、博士生导师,教育部长江学者特聘教授。研究方向为工业生物技术、代谢工程、生物转化和生物催化。在,、、、、、《生物工程学报》《微生物学报》等国内外生物工程类主流学术期刊上以第一或责任作者身份发表学术论文120余篇,其中SCI论文100余篇。出版科技著作2部。获得授权发明专利28项,申请或公开发明专利20余项。研究成果近5年来获国家技术发明二等奖、教育部科技进步一等奖、中国石油与化学工业联合会科技进步一等奖等5项科技奖励。

叶超, 徐楠, 陈修来, 等. 应用代谢网络模型解析工业微生物胞内代谢. 生物工程学报, 2019, 35(10): 1901–1913.
Ye C, Xu N, Chen XL, et al. Application of metabolic network model to analyze intracellular metabolism of industrial microorganisms. Chin J Biotech, 2019, 35(10): 1901–1913.
June 16, 2019;
August 19, 2019
Supported by:National Key Research and Development Program of China (No. 2018YFA0901401), the Scientific and Technological Innovation Leading Talents of National “Ten Thousand Talent Program” (2018), the National Natural Science Foundation of China (No. 21808083), the Key Technologies Research and Development Program of Jiangsu Province (Nos. BE2018623, BE2017622), the Key Field Research and Development Program of Guangdong Province (No. 2019B020218001).
Liming Liu. Tel/Fax: +86-510-85197875; E-mail: mingll@jiangnan.edu.cn
国家重点研发计划 (No. 2018YFA0901401),2018年“万人计划”科技创新领军人才支持经费,国家自然科学基金 (No. 21808083),江苏省科技支撑计划社会发展项目 (Nos. BE2018623, BE2017622),广东省重点领域研发计划项目 (No. 2019B020218001) 资助。
(本文责编 陈宏宇)
