Conserved sequences identify the closest living relatives of primates
2019-10-31MeiLingZhangMingLiLiAdeolaOluwakemiAyoolaRobertMurphyDongDongWuYongShao
Mei-Ling Zhang,Ming-Li Li,Adeola Oluwakemi Ayoola,Robert W.Murphy,Dong-Dong Wu,Yong Shao
1 Department of Acute Infectious Diseases Control and Prevention,Yunnan Provincial Centre for Disease Control and Prevention,Kunming Yunnan 650022,China
2 State Key Laboratory of Genetic Resources and Evolution,Kunming Institute of Zoology,Chinese Academy of Sciences,Kunming Yunnan 650223,China
3 Centre for Biodiversity and Conservation Biology,Royal Ontario Museum,Toronto M5S 2C6,Canada
4 Kunming College of Life Science,University of the Chinese Academy of Sciences,Kunming Yunnan 650223,China
ABSTRACT Elucidating the closest living relatives of extant primates is essential for fully understanding important biological processes related to the genomic and phenotypic evolution of primates,especially of humans. However, the phylogenetic placement of these primate relatives remains controversial, with three primary hypotheses currently espoused based on morphological and molecular evidence.In the present study,we used two algorithms to analyze differently partitioned genomic datasets consisting of 45.4 Mb of conserved non-coding elements and 393 kb of concatenated coding sequences to test these hypotheses. We assessed different genomic histories and compared with other molecular studies found solid support for colugos being the closest living relatives of primates.Our phylogeny showed Cercopithecinae to have low levels of nucleotide divergence,especially for Papionini,and gibbons to have a high rate of divergence. The MCMCtree comprehensively updated divergence dates of early evolution of Primatomorpha and Primates.
Keywords: Phylogeny; Colugos; Primates;Conserved non-coding elements;Divergence time
INTRODUCTION
The advancement of genomics has ushered an era in which genetic studies on the evolution of adaptively complicated traits have become possible.This is especially interesting for humans, non-human primates, and their closest relatives.However,such assessment requires a robust hypothesis of phylogenetic relationships.To date,the phylogeny of primates and their closest relatives remains a subject of intense debate. Analyses of different morphological or molecular datasets show conflicting topologies,most likely due to the accelerated evolution of the Euarchonta,which includes the Primates,Southeast Asian flying lemurs(Dermoptera),and tree shrews(Scandentia).Furthermore,apparent incomplete lineage sorting(ILS)can drive conflict between estimations of species trees and gene trees(Murphy et al.,2007;Nishihara et al.,2006;Scornavacca&Galtier,2017).
Massive parallel data have driven advancements in genomics and associated methods(Liu et al.,2010;Mirarab et al., 2014a, 2014b) and can be applied to hypothesize species trees.Analyses using a greater number of genes or diverse data can be used to parse credible species trees,such as that for modern birds (Jarvis et al., 2014).Longstanding hypotheses based on morphological and molecular proof have suggested a sister-group relationship between primates and tree shrews(Kay et al.,1992;Novacek,1992;Wible&Covert,1987).In addition,several other studies have regarded both tree shrews and flying lemurs(colugos)together as a sister group of primates(Bloch et al.,2007;Liu et al.,2001;Murphy et al.,2001;Nie et al.,2008;Sargis,2002; Springer et al., 2003, 2004). Further phylogenetic analysis incorporating paleontological evidence also suggests that primates and colugos are sister taxa (Beard, 1993).Previous molecular studies also support colugos as the closest living relatives of primates (Bininda-Emonds et al.,2007;Hudelot et al.,2003;Waddell et al.,2001).Genomic analyses further postulated a third potential topology:((primates, colugos), tree shrews) (Janecka et al., 2007;Perelman et al.,2011),though this was based on analyses of limited genomic changes(insertion and deletions,InDels)and few nuclear gene fragments.Furthermore,these analyses did not exclude biases due to ILS, data partitioning, or data insufficiency (Chojnowski et al., 2008; Patel et al., 2013;Rokas et al.,2003).
Whole genome data with a well-preserved evolutionary history for the genome of each species can be used to recover an exact species tree(Jarvis et al.,2014;Mitchell et al.,2015;Rokas et al.,2003;Wolf et al.,2002).Herein,we inferred the closest living relatives of primates and estimated the time scale for extant Euarchonta based on 23 existing genome datasets and an out-group(Mus musculus).Analyses relied on two types of data:393 kb of concatenated proteincoding sequences from 706 one-to-one orthologous genes(OOGs)and 45.4 Mb of concatenated conserved non-coding elements(CNEs).
MATERIALS AND METHODS
Identification of OOGs and sequence alignment/trimming analyses
We downloaded all protein-coding sequences of 20 species(primates,colugos,and tree shrews)(Table 1)from the NCBI assembly database(https://www.ncbi.nlm.nih.gov/).Data for Homo sapiens,Pan troglodytes,Tupaia belangeri,and Mus musculus were downloaded from Ensembl 88 (http://www.ensembl.org/info/data/ftp/). The dataset with the longest coding sequence(CDS)of each gene for each species was retained and then translated into proteins to detect paired OOGs using InParanoid-4.1(Remm et al.,2001)(parameters:$score_cutoff=40,$conf_cutoff=0.05,$group_overlap_cutoff=0.5,$seq_overlap_cutoff=0.5 and$segment_coverage_cutoff=0.25). Multiple species OOGs were captured by best reciprocal intersections. Prank v100802 (Loytynoja &Goldman,2005,2008,2010)(parameters:-f=fasta-F-codonnoxml-notree-nopost)and Gblocks v0.91b(Castresana,2000;Talavera&Castresana,2007)(parameters:-t=c-b4=5-b5=n)were used to align orthologous regions and to trim alignments with low-quality regions for each OOG,respectively.
Pairwise and multiple whole genome alignments
The pairwise whole genome alignments of Pan troglodytes,Gorilla gorilla, Macaca mulatta, Microcebus murinus,Galeopterus variegatus,and Mus musculus vs.Homo sapiens were downloaded from the University of California Santa Cruz(UCSC) pairwise alignments. We also downloaded repeatmasked whole genome sequences (17 species) from the NCBI assembly database, with the repeat-masked Homo sapiens genome obtained from the UCSC Genome Browser(http://genome.ucsc.edu/).Pairwise whole genome alignments for 17 species(Table 1)vs.Homo sapiens were obtained using LASTZ (Schwartz et al., 2003) with the following parameters:E=30,O=400,K=3 000,L=2 200,and M=50.A 24-way whole genome multiple alignment was then generated using Multiz v11.2 (Blanchette et al., 2004) with default parameters, and with Homo sapiens regarded as the reference taxon.
Detection of CNEs and pre-processing
Phastcons(Siepel et al.,2005)was utilized to obtain elements with conserved scores under a given multiple whole genome alignment file and phylo-HMM. The phylo-HMM analysis assumed a conserved and non-conserved state.Modifications of parameters were:--target-coverage 0.3,--expected-length 45,and--rho 0.31.The phylogenetic model for non-conserved regions was produced by phyloFit in the PHAST package(Hubisz et al., 2011) (http://compgen.cshl.edu/phast/). We then used an in-house Python script to extract the CNEs from multiple sequence alignments across multiple genomes.Our screening required that the CNEs fulfilled three criteria:(1)alignments with gaps in at least one of the 24 species were deleted in this study;(2)multiple sequence alignments overlapped with conserved non-coding regions;and(3)length of overlapping sequences exceeded 10 bp.
Estimates of phylogeny, genomic divergence, and divergence time
Codon positions 1,2,and 1+2 of each filtered OOG among the 24 species were extracted and concatenated.All CNEs were concatenated and trimmed using Gblocks(Castresana,2000;Talavera&Castresana,2007).The maximum likelihood(ML)trees using the concatenated genes were reconstructed using RAxML v8.1.17 (Stamatakis, 2014) with the GTR+GAMMA substitution model and 1 000 bootstraps.We used 1 000 rapid bootstrap replicates to assess branch reliability.Modeltest(Posada&Crandall,1998)was selected to detect the best substitution model and MrBayes v3.2.6(Huelsenbeck& Ronquist, 2001) was used to reconstruct a Bayesian inference(BI)tree.The chain length was set to 10 000 000(10 000 generation/sample),with the first 100 000 samples discarded as burn-in. The reconstructed single-gene trees with a root(M.musculus)from RAxML v8.1.17(Stamatakis,2014) were applied using maximum pseudo-likelihood estimation of the species tree(MP-EST)(Liu et al.,2010;Shaw et al., 2013) with default parameters to estimate a species tree based on a multispecies coalescent model.This approach was assumed to resolve conflicts between concatenated and coalescent species trees if ILS existed in our dataset. Relative genomic divergence (nucleotidesubstitutions/site) was visualized as branch lengths in the concatenated species phylogeny.
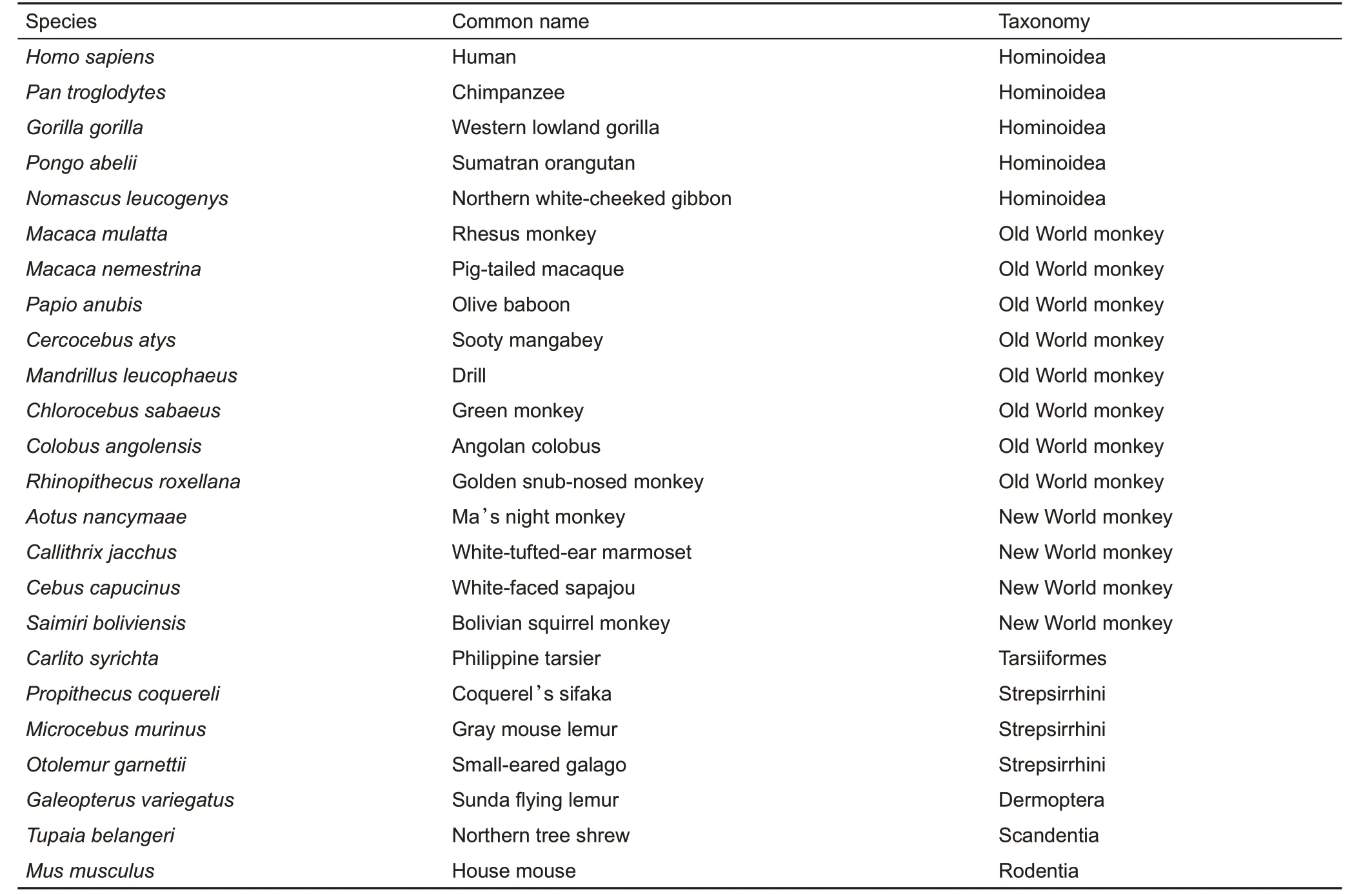
Table 1 List of taxa,including primates,colugos,tree shrews,and rodents,used in our analyses
We estimated divergence time using MCMCtree(dos Reis&Yang,2011;Yang,2007),with five fossil calibration points used to determine approximate likelihood calculations(Fan et al.,2013;Franzen et al.,2009;Matsui et al.,2009;Poux&Douzery,2004;Vignaud et al.,2002).Using MCMCtree(dos Reis&Yang,2011;Yang,2007),we obtained 10 000 trees with a sampling frequency of 50 and burn-in of the first 10 000 iterations.The parameters were modified as follows:'clock=3','model=0','alpha=0','ncatG=5','cleandata=1','BDparas=1 1 0','kappa_gamma=6 2','alpha_gamma=1 1','rgene_gamma=2 2','sigma2_gamma=1 10',and'finetune=1:.00356 0.02243 0.00633 0.12.43455'.All other parameters were set to default.Tracer(http://beast.bio.ed.ac.uk/Tracer)was used to detect convergence.
RESULTS AND DISCUSSION
Coding data and phylogeny based on coding partitioned models
We identified 738 OOGs among the 24 species(Table 1).After multiple sequence alignments and removal of ambiguous sites/regions for each gene, 706 OOGs were retained. Because rapidly evolving nucleotide sites are phylogenetically less informative than slowly evolving ones,especially for ancient groups (Kälersjö et al., 1999), we deleted the third codon positions for each OOG. The remaining data were concatenated and partitioned by codon position as follows:codon position 1(196 501 bp),position 2(196 501 bp), and positions 1+2 (393 002 bp).We employed RAxML v8.1.17(Stamatakis,2014)to determine phylogeny based on ML algorithm with using the GTR+GAMMA substitution model. The topologies of the three consensus trees potentially resolved the deep-branch phylogeny of primates. The representative colugos constituted a sister group of primates, which together formed the Primatomorpha.Compared to the concatenated codon 1 and codon 2 sequences,the concatenated codons 1+2 provided greater support for all nodes.Only one node did not have 100%bootstrap support(BS)(Figure 1).The BI analysis was consistent with the sequence data of the concatenated codons 1+2, with high Bayesian posterior probability(BPP)for each node.
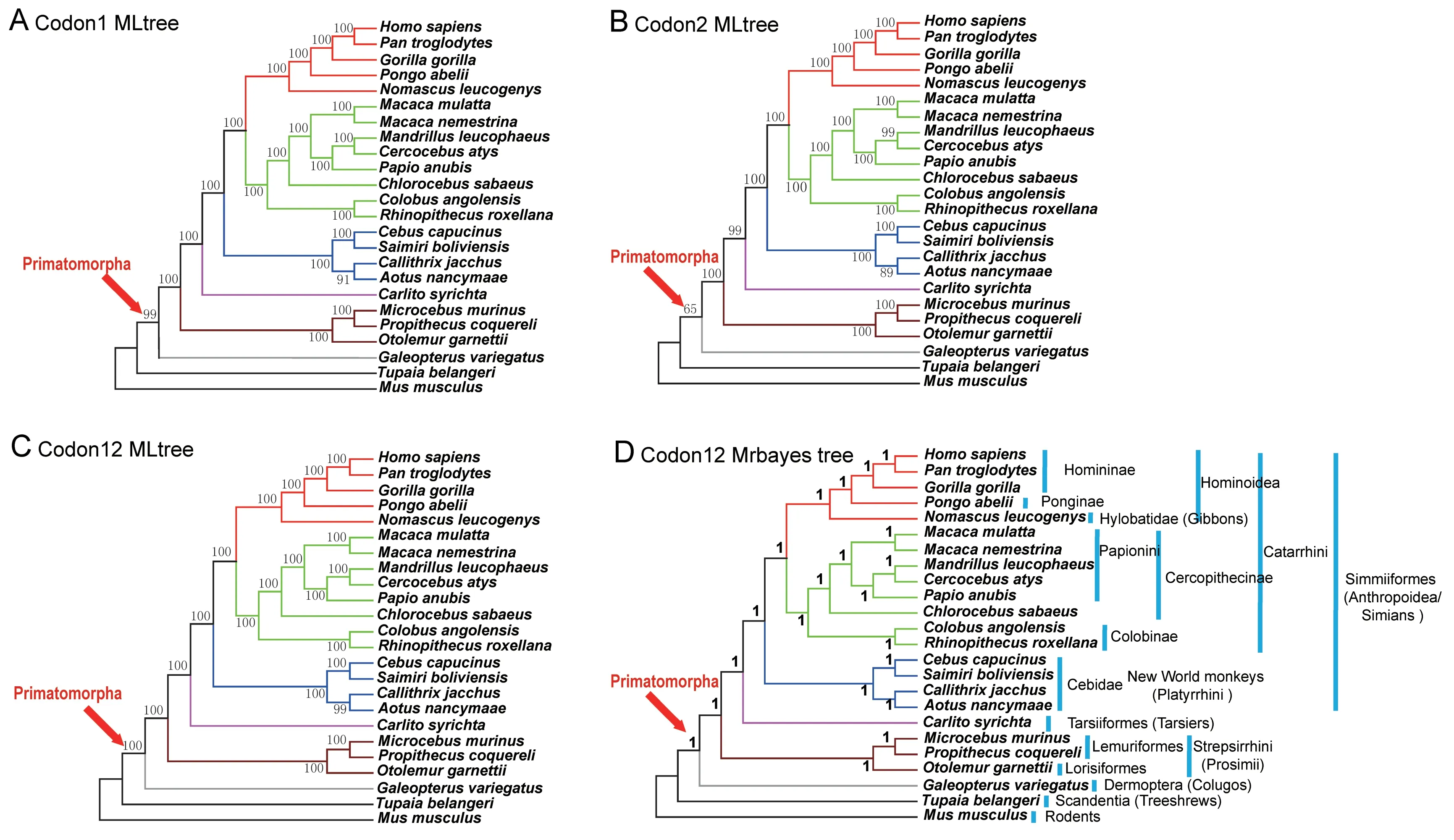
Figure 1 Reconstructed phylogenies based on concatenated sequences from first, second, and first+second codon positions,respectively,of 706 OOGs
Phylogenetic analyses of the three data-partitions were concordant with the ML and BI topologies for Primatomorpha.However,complicated biological processes,such as ILS,can influence the reconstruction of species trees as different protein-coding genes may have divergent evolutionary histories during the early diversification of Euarchonta(Murphy et al., 2007; Nishihara et al., 2006). Thus,concatenated gene sequences may result in a false-positive species tree with higher nodal BS or BPP(Kubatko&Degnan,2007).Thus,we estimated the best topology of each gene tree based on the ML algorithm across 24 species, and discovered that discordance between the gene and species trees indeed existed in protein-coding genes in our dataset,which included 100 gene trees (14.1%) supporting tree shrews as the closest living relatives of primates and 25 gene trees(3.5%)supporting both tree shrews and colugos as a sister group of primates. However, we identified a higher proportion of genes(130 genes,~18.4%)supporting colugos as the closest living relatives of primates (Supplementary Figure S1). Multispecies coalescence can resolve discordance between gene and species trees,even in the presence of ILS(Mirarab et al.,2014a;Song et al.,2012;Zhong et al.,2013).Therefore,we used MP-EST(Liu et al.,2010;Shaw et al.,2013)to construct a coalescent phylogeny.The method maximizes a pseudo-likelihood function to infer a species tree with 1 000 bootstrap replicates.The topology of the coalescent species tree was concordant with the one using the concatenated method.The deep node at the split of colugos and primates had moderate BS(77%)and the node for the last common ancestor of Callithrix jacchus and Aotus nancymaae had low BS(55.6%)(Supplementary Figure S2).For the phylogeny of primates, the species tree topology based on the concatenated data was consistent with that of the coalescent data.
Species tree based on conserved non-coding elements
Protein-coding regions only reflect a part of a genome's evolutionary story.Most genome sequences are from noncoding regions.Previous studies have shown that CNEs have important biological functions, including development and transcriptional regulation(Mikkelsen et al.,2007;Woolfe et al.,2005).We used LASTZ(Schwartz et al.,2003)to perform pairwise genome alignments with human as the reference,and then used MULTIZ v11.2 (Blanchette et al., 2004) to obtain whole genome alignments across the 24 species.Phastcons (Siepel et al., 2005) identified 215.6 Mb of concatenated CNEs,with Gblocks v0.91b(Castresana,2000;Talavera&Castresana,2007)(parameters:-t=d-b4=5-b5=n)then used to trim the data for high quality.These analyses obtained 45.4 Mb of data across all 24 species.The RAxML v8.1.17(Stamatakis,2014)analyses of the CNEs produced a phylogeny using the GTR+GAMMA substitution model and 1 000 bootstrap replicates.Each of the nodes for the potential species tree arrived at a 100%BS(Figure 2A).This analysis also supported the hypothesis that colugos are the closest living relative of primates.The tree depicted a very short branch length from the last common ancestor of primates and colugos, suggesting that the last common ancestor experienced rapid radiation.This discovery may explain,at least in part,why the sister group of primates is controversial.Analyses using mitochondrial and nuclear sequences did not obtain this result(Murphy et al.,2001;Springer et al.,2003),whereas the globally concatenated data from codons 1+2 plus CNEs inferred a consistent species tree (Supplementary Figure S3).
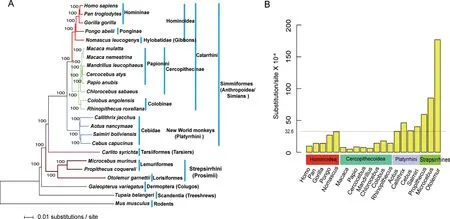
Figure 2 Maximum likelihood tree(ML)and genomic divergences based on conserved non-coding elements(CNEs)
CNEs and coding trees vs.InDels and nuclear gene trees
Compared to limited morphological, mitochondrial, and nuclear gene assessments,high-quality whole genomes and robust algorithms can identify, alter, and extend previous taxonomic conclusions. Our phylogenetic analyses, rooted using a rodent(Mus musculus),covered the main branches of primates for which genomes exist.Many factors,such as gene duplication, admixture, introgression, hybridization,recombination, convergent evolution, and natural selection,can influence the reconstruction of a true species tree(Degnan & Rosenberg, 2009; Rokas et al., 2003;Scornavacca&Galtier,2017).However,ILS has often been invoked to explain conflicts between species and gene trees.Many phylogenetic studies examining conflicting signals of different genes have reported considerable discordance across gene trees,e.g.,in fruit flies(Pollard et al.,2006)and finches(Jennings&Edwards,2005).Here,our concatenated and coalescent models supported the hypothesis that colugos are the extant sister group of primates,which together form the Primatomorpha. Concatenated sequences have potentially more informative sites.The coalescent model fills in discordances between the estimated species tree and gene tree once ILS occurs.All methods of analyses in the current study considered different data partitions (protein-coding genes and non-coding sequences), but still obtained a consistent species tree.Our results were in concordance with the hypothesis that colugos are the closest living relatives of primates but refuted the hypotheses that tree shrews or tree shrews plus colugos are the sister group of primates.These observations provide similar insight as two classical molecular studies,which utilized InDels in genomes(Janecka et al.,2007)and 54 sequenced nuclear genes to infer the primate species tree(Perelman et al.,2011).However,our analyses still cannot exclude important influences of many other complex evolutionary histories, such as ancient admixture,introgression, or other potential factors. Recent lines of evidence have indicated that ancient gene flows may potentially underpin phylogenetic discordances(de Manuel et al.,2016;Kuhlwilm et al.,2019;Li et al.,2016;Melo-Ferreira et al.,2014;Nater et al.,2017;Rogers et al.,2019;Tung&Barreiro,2017;Wu et al.,2018).Therefore,future population genomic studies with deeper coverage and advanced methods need to validate this possibility in clades.
Estimates of genomic divergence in primates
Primate phylogeny can be used to estimate the relative levels of genomic divergences based on branch lengths.The branch uniting Homininae and Pongo abelii separated them from Nomascus leucogenys,which was assigned to Hylobatidae.This short branch length indicated that Hominoidea radiated rapidly as ((Ponginae, Homininae), Hylobatidae). Overall,primate genomes have varying levels of genomic divergences. According to previous studies, gibbons(Hylobatidae) have an extremely high proportion of chromosomal rearrangement(Jauch et al.,1992;Muller et al.,2003; Perelman et al., 2011). Our analyses indicated that Nomascus leucogenys had the highest rate of genomic divergence (32.3 substitutions/site×10-4) among species of Hominoidea. This was almost equivalent to the mean nucleotide divergence (32.6 substitutions/site×10-4) of all primate genomes(Figure 2B).The results of a previous study(Perelman et al.,2011)and our research suggest that this may be a consequence of genomic rearrangements within Nomascus leucogenys; however, this needs additional investigation. Our results also showed that Old World monkeys had the lowest genomic divergence, and this contrasted with their sister group, Hominoidea and other primates. Low levels of divergence were specific characteristics of Papionini, including Macaca mulatta,Macaca nemestrina, Mandrillus leucophaeus, Cercocebus atys, and Papio anubis. Speciation patterns of Old World monkeys are remarkably misleading due to convergent morphological traits,behavior,and reproduction,as well as sympatric hybridization (Perelman et al., 2011). Here, the rapid speciation and radiations in Papionini may have involved in reticulate evolution via natural hybridization (Arnold &Meyer,2006).
Early evolutionary dates of Primatomorpha and Primates
The timing and early evolution of Primatomorpha and Primates are intriguing.A MCMCtree approach(dos Reis&Yang,2011;Yang,2007)with five fossil constraints(Fan et al.,2013; Franzen et al., 2009; Matsui et al., 2009; Poux &Douzery,2004;Vignaud et al.,2002)was used to estimate evolutionary dating(Figure 3).According to previous methods(Liu et al.,2017;Reis et al.,2018),we generated a plot of the posterior distributions to estimate the accuracy of our divergence dating analyses for calibrated nodes,compared to a previous primate phylogeny study(Perelman et al.,2011)and TimeTree (http://www.timetree.org/) (Supplementary Figure S4).Our analyses were consistent with their results and thus ensured that our analyses were reliable.Our results suggested the origin of Primatomorpha at ~88.0 million years ago(Mya),Primates at ~77.4 Mya,Strepsirrhini at ~70.6 Mya,and Tarsiiformes at ~65.0 Mya(Figure 3).Previously,groups at the base of Euarchonta have been difficult to resolve due to the rapid radiation of ancestral lineages before the Cretaceous-Tertiary(K/T)boundary(~65 Ma)(Wible et al.,2007)and limited sampling of genes and taxa(Janecka et al.,2007;Springer et al.,2003).The estimated dates of origins of Primates, Strepsirrhini, and Tarsiiformes are similar to previous inferences based on nuclear and mitochondrial genes (Jameson et al., 2011; Pozzi et al., 2014). This consistency suggests that our estimates are potentially reliable.These events predated the K/T boundary(Wible et al.,2007).Short branch lengths occurred for the last common ancestor of colugos and primates,as well as for Tarsiiformes and Simiiformes in the CNE tree (Figure 2A). Such rapid divergences may explain why some morphological and molecular studies(Bloch et al.,2007;Sargis,2002)fail to resolve the placements of colugos and tarsiers.Analyses also estimated that extant strepsirrhines arose ~53.2 Mya,based on three species(Propithecus coquereli,Microcebus murinus and Otolemur garnettii),which covered two main groups of Strepsirrhini:Lemuriformes and Lorisiformes.
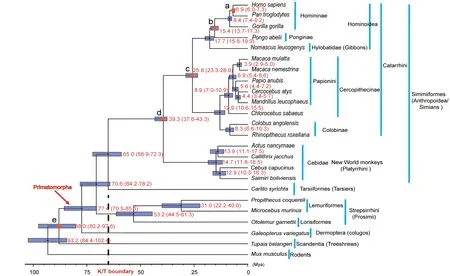
Figure 3 Estimated divergence dates of Euarchonta
CONCLUSIONS
Our analyses provide a potentially robust assessment of the closest living relatives and divergence times of living primates.The improved conserved non-coding elements and partitioned protein-coding sequences will facilitate further comprehensive studies into the biological processes related to the genomic and phenotypic evolution of primates,especially of humans.Furthermore,our analyses provide a useful resource for the phylogenetic reconstruction of ancient groups.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS'CONTRIBUTIONS
Y.S.and D.D.W.conceptualized the work and revised the paper.M.L.Z.conducted bioinformatic analysis and drafted the paper.M.L.L.analyzed data.A.O.A.and R.W.M.revised the paper.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We acknowledge Wei-Wei Zhou from Kunming Institute of Zoology,CAS for his comments in phylogenetic analyses and estimates of divergence time.
杂志排行

Zoological Research的其它文章
- From our roots,we grow Celebrating the 60th anniversary of the Kunming Institute of Zoology,Chinese Academy of Sciences
- Genomic insights into ruminant evolution:from past to future prospects
- Antimicrobial peptides:new hope in the war against multidrug resistance
- Chromosomal level assembly and population sequencing of the Chinese tree shrew genome
- Superior intestinal integrity and limited microbial translocation are associated with lower immune activation in SIVmac239-infected northern pig-tailed macaques(Macaca leonina)
- Systematic relationships of Chinese freshwater semisulcospirids(Gastropoda,Cerithioidea)revealed by mitochondrial sequences