Genomic insights into ruminant evolution:from past to future prospects
2019-10-31BaoWangLeiChenWenWang
Bao Wang ,Lei Chen,Wen Wang
1 State Key Laboratory of Genetic Resources and Evolution,Kunming Institute of Zoology,Chinese Academy of Sciences,Kunming Yunnan 650223,China
2 Kunming College of Life Science,University of Chinese Academy of Sciences,Kunming Yunnan 650204,China
3 Center for Ecological and Environmental Sciences,Northwestern Polytechnical University,Xi'an Shaanxi 710072,China
4 Center for Excellence in Animal Evolution and Genetics,Chinese Academy of Sciences,Kunming Yunnan 650223,China
ABSTRACT Ruminants (Ruminantia) are among the most successful herbivorous mammals, exhibiting wideranging morphological and ecological characteristics(such as headgear and multichambered stomach)and including various key livestock species(e.g.,cattle,buffalo,yak,sheep,and goat).Understanding their evolution is of great significance not only in scientific research but also in applications potential for human society. The rapid growth of genomic resources provides unprecedented opportunities to dissect the evolutionary histories and molecular mechanisms underlying the distinct characteristics of ruminants. Here we summarize our current understanding of the genetic, morphological, and ecological diversity of ruminants and provide prospects for future studies.
Keywords: Ruminantia; Genome evolution;Phylogenomics;Traits;Adaptive evolution
INTRODUCTION
Ruminantia mammals are among the most successful herbivores, and include six families: i.e., Antilocapridae,Bovidae, Cervidae, Giraffidae, Moschidae, and Tragulidae.Ruminantia comprises at least 200 extant species, with Bovidae the most species-rich, consisting of at least 143 species(Heller et al.,2013),including important livestock such as cattle (Bos taurus), yak (Bos grunniens), sheep (Ovis aries), and goat (Capra hircus). Ruminants are distributed across extensive habitats,including different latitudes(from tropical to Arctic regions),different altitudes(from plains to plateaus),and different ecological environments(from deserts to rainforests). Moreover, ruminants exhibit several distinct anatomical features,such as headgear and multichambered stomach.Compared with other herbivores,such as horses,the rumen and omasum in ruminants enable more efficient utilization of plant cellulose(Clauss&Rössner,2014;Janis,1976;Russell&Rychlik,2001).This efficient procurement of food energy may be an important reason why ruminants are so prosperous.Ruminants also possess specialized dentition,speed,and considerable variation in body size.Furthermore,they have played an important role in human civilization due to the domestication of several species, such as cattle,buffalo, yak, sheep, and goat. In modern society, these domestic animals remain important for human diet and utilization.Therefore,understanding the evolution and origin of ruminants is of great importance for scientific research and for application prospects in human society.
In the past decade, we have witnessed the rapid development of genome sequencing technologies and a resultant wealth of knowledge from animal genomes.To date,the genomes of 73 ruminant species have been reported(Table 1), with increasing availability of resequencing and transcriptome data for representative species, like cattle(Chen et al.,2018;Daetwyler et al.,2014;Medugorac et al.,2017;Soubrier et al.,2016),goat(Alberto et al.,2018;Wang et al.,2016),sheep(Alberto et al.,2018;Hu et al.,2019;Naval-Sanchez et al.,2018),and yak(Qiu et al.,2015;Wang et al.,2019a).Most previous studies have only focused on the evolution of individual species by decoding individual genomes and utilizing large-scale resequencing data;e.g.,high-altitude adaptation studies on yak and Tibetan antelope(Ge et al.,2013;Qiu et al.,2012;Qiu et al.,2015).Nowadays,large-scale sequencing studies of multiple species across high classification orders have provided extraordinary biological discoveries,e.g.,avian phylogenomics(Jarvis et al.,2014).We recently launched the Ruminant Genome Project and conducted a large-scale genomic study of ruminants based on integrated analysis of 51 ruminant genomes(44 newly assembled genomes and seven public genomes)and corresponding transcriptome, resequencing, and functional experiments(Chen et al.,2019;Lin et al.,2019;Wang et al.,2019b). The availability of large-scale genomic resources should provide new insights into our understanding of ruminant diversity, evolution, biogeographic patterns, and adaptation mechanisms.
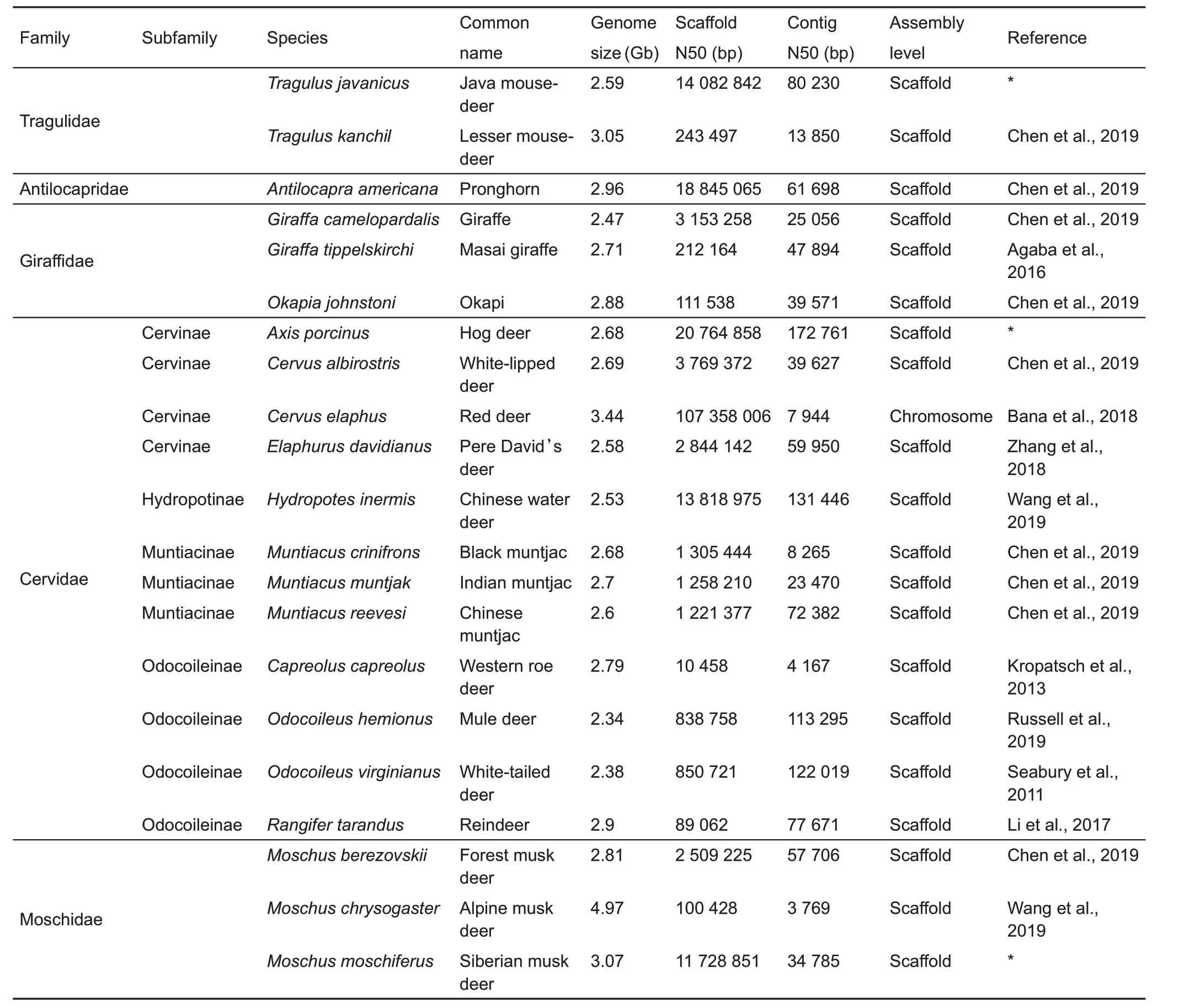
Table 1 Currently available genome assemblies for ruminant species
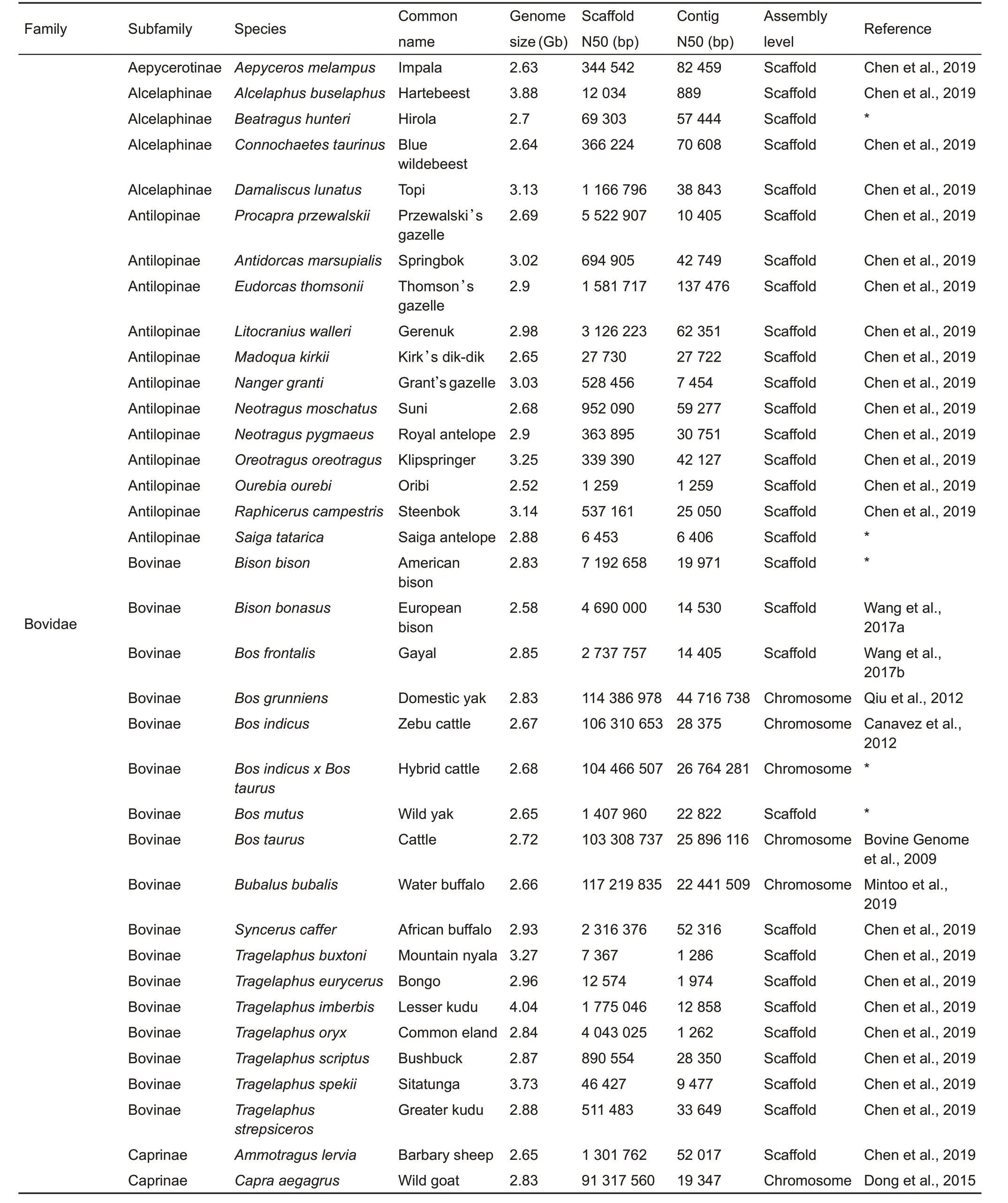
Continued
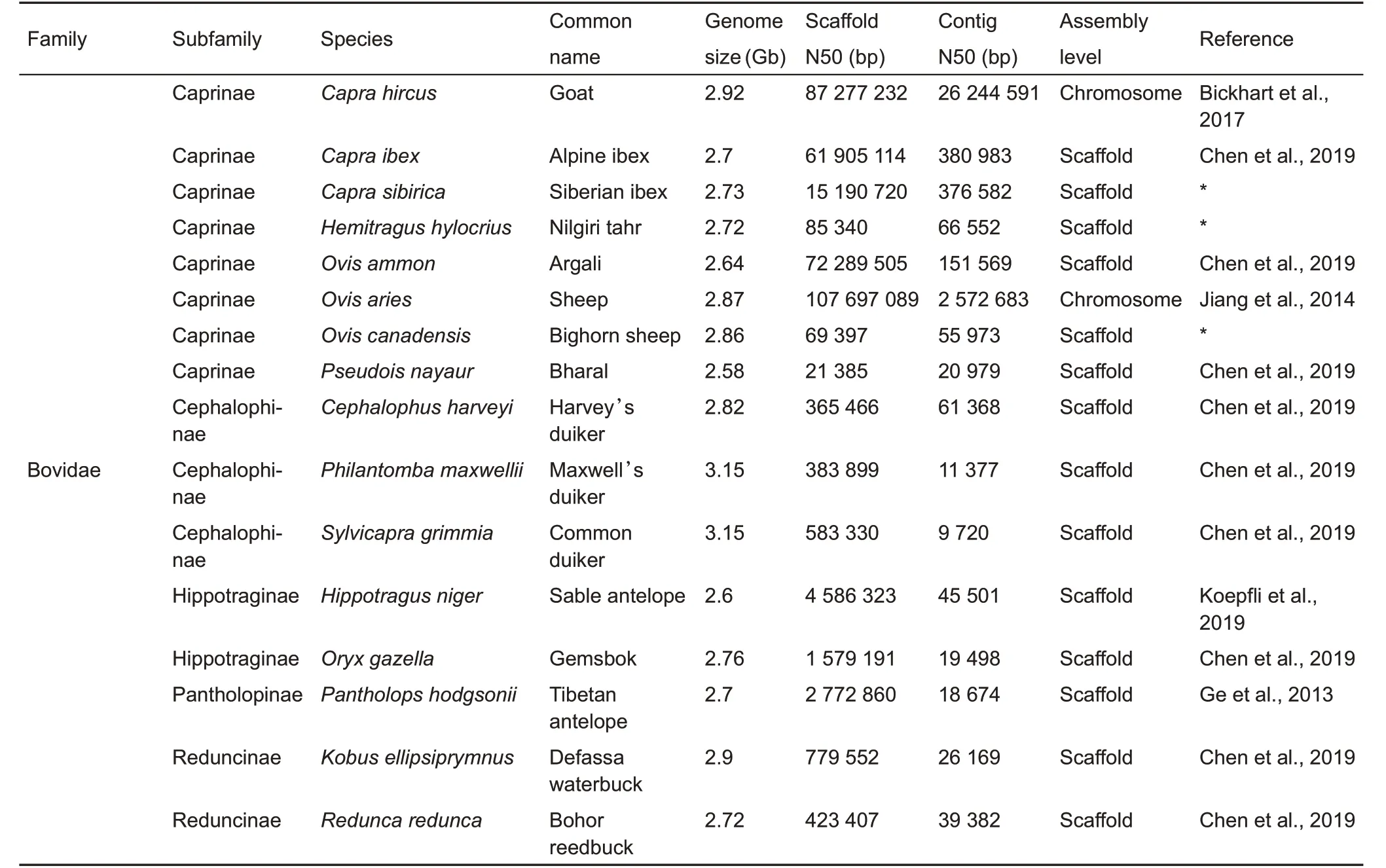
Continued
In this review, we first summarize current knowledge of ruminant evolution learned from the genomic era,especially phylogenetic relationships, genetic basis and evolution of complex traits,genetic basis underlying local adaptations,and domestication of livestock ruminants. We then provide an outlook for future research on ruminants given the rapid advances in the accumulation of genomic resources and increasing efforts in functional genomics.
PHYLOGENETIC RELATIONSHIPS OF RUMINANTS
Phylogeny is essential to biology not only because it reflects the evolutionary history of species or lineages and divergent events,but also provides a framework for tracing the evolution of distinct complex characteristics. In previous studies,phylogenetic trees of ruminants were constructed based on morphological data(e.g.,teeth and skeletons),fragments of nuclear DNA, or complete mitochondria DNA (Bibi, 2013;Decker et al.,2009;Gatesy et al.,1992;Hassanin&Douzery,2003;Hassanin et al.,2012).However,controversies in these phylogenetic trees remain,resulting in different placements at the genus,subfamily,and even family level.To resolve these issues,Chen et al.(2019)provided a robust phylogenetic tree based on whole genome data from 51 ruminants,clarifying most of the controversies in ruminant phylogeny.Here,using all available genomes(Table 1),we reconstructed a simplified phylogenetic tree (Figure 1). In previous studies,Antilocapridae was considered an outgroup to all other pecorans (all ruminants, excluding Tragulidae) based on mtDNA (Bibi, 2013; Hassanin et al., 2012; Hernández Fernández&Vrba,2005).Moschidae was placed at the base of Pecora (Janis, 1987; Janis & Theodor, 2014; Webb &Taylor, 1980) or as a sister group to Bovidae based on morphological data(Sánchez et al.,2010),or proposed as a sister group of Cervidae or Bovidae based on mtDNA(Bibi,2014; Dos Reis et al., 2012; Hassanin & Douzery, 2003;Hassanin et al.,2012).The whole genome tree confirms the sister-group relationship between Antilocapridae and Giraffidae,and Moschidae as a sister group with Bovidae.In addition, some controversies in Bovidae (Hernández Fernández&Vrba,2005)are also resolved,with Reduncinae confirmed as a sister group of Caprinae,Alcelaphinae,and Hippotraginae.
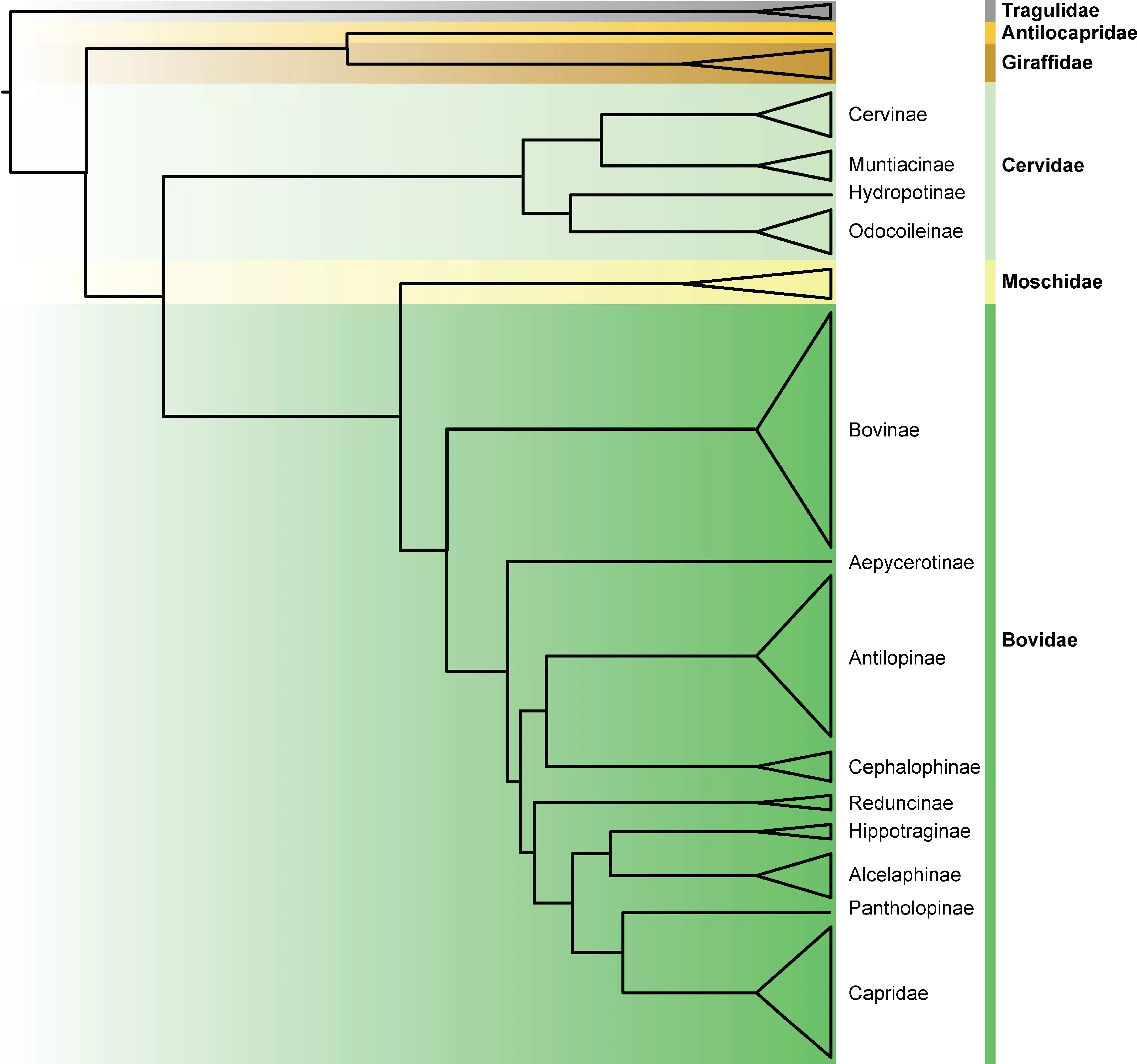
Figure 1 Phylogenetic relationship among ruminants
The above phylogenic controversies may be due to incomplete linage sorting(ILS)or to introgression between linages.A high level of phylogenetic heterogeneity is observed at the family level of ruminants,showing that only 21.5%of sample genomic windows are consistent with the topology of the whole genome tree (Chen et al., 2019). These phylogenetic disagreements can primarily be explained by ILS,resulting from the rapid radiation of ruminant families during the Miocene(Chen et al.,2019).In addition,a strong gene flow signal has been reported among Giraffidae,Cervidae, Bovidae, and Moschidae (Chen et al., 2019),implying ancestral introgressions may also partially account for the phylogenic debates. In particular, pervasive introgression among Bos species has been reported from other studies(Wang et al.,2018a;Wu et al.,2018),which provides support for introgression effects on the phylogenetic relationships of ruminants.
EVOLUTION OF IMPORTANT TRAITS
Most traits are consequences of long-term evolution and form the basis of biodiversity. As one of the most successful mammalian linages,ruminants have evolved extensive and unique morphological traits, such as headgear,multichambered stomach,substantial variation in body size,cold (Arctic) adaptation (reindeer), and high-altitude adaptation. The availability of large-scale genomic data,together with transcriptomics and functional genomics,has enabled investigations into the genetic basis and evolution of complex traits in ruminants.
Evolution of headgear
Ruminants are the only mammalian group with osseous headgear(Davis et al.,2011).Headgear is not only used in self-defense against predators but also in intraspecies competition for mates and territories (Bubenik & Bubenik,2012;Davis et al.,2011).Four families of ruminants have headgear,including Giraffidae,Antilocapridae,Cervidae,and Bovidae. Headgear in different families exhibits distinct morphological characteristics,such as ossicones in giraffids,pronged horns in pronghorns,antlers in cervids,and horns in bovids(Bubenik&Bubenik,2012).The genetic mechanisms involved in headgear have long been a topic of interest to evolutionary biologists(Aristotle,1991;Goss,1983).
The expression profiles, phylogenies, and convergent headgear losses support a single evolutionary origin of ruminant headgear(Chen et al.,2019;Wang et al.,2019b).The highly or specially expressed genes in horns and antlers are mostly co-expressed in bone, skin, nerve tissue, and testis,and many positively selected genes are associated with conserved elements involved in neural functions(Wang et al.,2019b). The headgear in ruminants likely originated from neural crest stem cells,which diverged into horns,antlers,ossicones,and pronged horns in the bovids,cervids,giraffids,and pronghorns, respectively. The loss of headgear in Moschidae and Hydropotinae was likely due to convergent pseudogenization of the gene RXFP2(Wang et al.,2019b).
Interestingly,antlers in the Cervidae have evolved further remarkable features. Professor Richard Goss from the University of Brown wrote, "The antlers of deer are so improbable that if they had not evolved in the first place they would never have been conceived even in the wildest fantasies of the most imaginative biologists"(Goss,1983).Deer antlers are the only completely regenerative organ found in mammals.They exhibit extremely rapid growth rates(~1.7 cm/day in red deer)that can even surpass cancerous tissue growth in cell proliferation (Goss, 1983), although deer experience low rates of cancer (Griner, 1983; Lombard &Witte,1959).Wang et al.(2019b)showed that antler-specific expressed genes are enriched in the axon guidance pathway,suggesting that antler growth may be involved in neural processes.Strikingly,their results also revealed that protooncogenes (FOS, REL, FAM83A) and tumor suppression genes are positively selected in cervids,thus explaining the high growth rate of deer antlers but low cancer rate in cervids.Understanding the genetic basis of rapid antler regeneration could provide new insights into the regeneration of mammalian organs and oncogenesis.
Evolution of multichambered stomach
The ruminant digestive system plays important ecological and functional roles in evolution.The appearance of the rumen and its close interaction with microorganisms allows ruminants to gain energy efficiently,thus providing unique evolutionary advantages and promoting the prosperity and diversity of ruminant groups.Compared with non-ruminants(e.g.,horses),ruminants can achieve higher utilization of plant fiber due to their unique forestomach fermentation system(Janis,1976).Most previous studies have focused on the interaction between microorganisms and the rumen (Bovine Genome Sequencing and Analysis Consortium et al.,2009;Seshadri et al.,2018;Stewart et al.,2018;Zhang et al.,2016).However,little is known about the origin and evolution of the multichambered stomach.
To investigate the origin and evolution of the ruminant stomach, Chen et al. (2019) compared gene expression profiles from 516 samples covering 50 tissues of sheep,together with large-scale genomic data. The expression profiles suggested that the rumen,reticulum,and omasum may have originated from the esophagus,as also suggested by Warner (1958) and Xiang et al. (2016), whereas the abomasum was closer to the intestine.Newly evolved genes also seem to have played an important role in the evolution of the rumen. Seven newly evolved genes identified at the ancestor of ruminants are specially expressed in the rumen,two of which,PRD-SPRRII and TCHHL2,are structural genes(Chen et al.,2019;Jiang et al.,2014).PRD-SPRRII is related to the cornification of the keratin-rich surface of the rumen,and TCHHL2 plays a role in cross-linking keratins at the rumen surface(Jiang et al.,2014).The omasum is a newly evolved organ in pecorans,resembling the rumen in structure and function (Millen et al., 2016). Among the 75 genes specifically and highly expressed in the omasum compared with other organs, one gene is newly evolved(LOC101107119) and another (SCNN1D) exhibits pecoranspecific amino acid changes(Chen et al.,2019).In addition,four genes(i.e.,SIM2,PAX9,KCNK5,and DENND2C)show pecoran-specific conserved non-exonic elements (CNEs)within their immediate upstream/downstream 10 kb regions(Chen et al., 2019). Based on gene family analysis, the lysozyme c family is also expanded in ruminants,containing ten or more copies(Chen et al.,2019;Irwin,2015),whereas other outgroup mammals have only one or a few copies.Furthermore, most expanded copies of lysozyme c are expressed in the abomasum, suggesting adaptation to its microbe-rich environment.
Evolution of body size
Ruminants exhibit extensive variation in body size,from 2 kg(e.g.,lesser mouse deer,Tragulus kanchil)to 1 200 kg(e.g.,giraffe,Giraffa camelopardalis and African buffalo,Syncerus caffer)(Castelló,2016).At the family level,body size is largest in Giraffidae,followed by medium-sized Antilocapridae and Cervidae, and relatively small-sized Tragulidae and Moschidae(Clauss et al.,2003).Bovidae exhibits varied body size, with Bovinae generally being the largest (Castelló,2016).Ruminants living in open grasslands also tend to be larger,which can help in resisting predators and adapting to changes in the environment.Ruminants living in the jungle tend to be smaller in size, which allows for more flexible movement and thus predator avoidance.
Genes associated with body size have significantly higher non-synonymous substitution rates in large-and small-sized bovid species(Chen et al.,2019).Specifically,six genes(i.e.,CXCL13,RNF115,NPNT,KL,SLC9A3R1,and MSTN)are associated with increased body size and five genes(i.e.,SBDS,BMP3,LRRN3,NFATC3,and SMARCAL1)are related to reduced body size via regulation of bone and muscle development. Several genes also have functional support from other studies.For example,SLC9A3R1 plays a role in osteogenesis by mineralizing osteoblasts,with disruption of this gene resulting in reduced body weight in mice(Shenolikar et al., 2002). MSTN regulates muscle cell growth and differentiation(Sartori et al.,2013),and mutations in MSTN can affect muscle mass in livestock species such goat,sheep,and cattle(Grobet et al.,1997;Li et al.,2016;Luo et al.,2014;Wang et al.,2018b)and other mammals(Bi et al.,2016;Gu et al.,2016).SBDS serves an important role in cell proliferation and is a causal gene of Shwachman-Diamond syndrome in humans(Boocock et al.,2003),which is characterized by short stature and skeletal abnormalities. For individual species,the special stature of giraffes enables access to food and is an adaptation to tropical grassland. Those genes associated with bone development with giraffe-specific mutations are involved in the transforming growth factor-b(TGF-b),Hedgehog,Notch,Wnt,and FGF signaling pathways(Agaba et al.,2016;Chen et al.,2019).
Reindeer adaptation to Arctic regions
Reindeer(Rangifer tarandus)are naturally distributed across Arctic and sub-Arctic regions, and consequently face numerous challenges, including severe cold, limited food availability,and prolonged periods of light or darkness(Blix,2016).To adapt to these environments,reindeer have evolved several strategies,including special lipid metabolism to avoid heat loss and to induce circadian arrhythmicity for long periods of light or dark(Lu et al.,2010;Stokkan et al.,2007;Van Oort et al., 2005). Based on detailed comparative genomic analysis,Lin et al.(2019)identified two positively selected genes(i.e.,CYP27B1 and POR)involved in the vitamin D metabolism pathway.Functional experiments have also shown that enzymes encoded by these two genes exhibit much higher catalytic activity than that of their orthologs in goat and roe deer(Lin et al.,2019).Eight genes with reindeerspecific mutations have been reported to be involved in the circadian rhythm pathway, with four being rapidly evolving genes.Among these genes,Pro1172Thr(P1172T)mutation in PER2 results in loss of binding ability with CRY1,which can cause arrhythmicity(Lin et al.,2019).
Adaptation to high altitude
Ruminants are distributed across a wide range of habitats,including high-altitude regions such as the Tibetan Plateau,with risks of hypobaric hypoxia.These ruminants include the yak,Tibetan sheep,and Tibetan goat,which are three key livestock species serving as sources of meat and fiber for Tibetan inhabitants(Hu et al.,2019;Qiu et al.,2012;Wang et al., 2016). In addition, the Tibetan antelope (Pantholops hodgsonii)and Marco Polo sheep(Ovis ammon polii)also exhibit adaptation to high altitude(Ge et al.,2013;Yang et al.,2017). Therefore, understanding the genetic basis and mechanisms underlying high-altitude adaptation is of great value in many scientific fields.
Genetic variation related to transcription factors, oxygen sensors, and target genes has been exploited to detect signals of selection in ruminant species such as the Tibetan sheep(Hu et al.,2019),Tibetan goat(Wang et al.,2016),yak(Qiu et al.,2012),and Tibetan antelope(Ge et al.,2013).In yak,three genes,identified as positively selected in response to hypoxia,are involved in hypoxia-inducible factor-1α(Hif-1α),including two important regulators(Adam17 and Arg2)and one target gene(Mmp3)(Qiu et al.,2012).In Marco Polo sheep,several genes associated with hypoxia response have been identified,including ryanodine receptor 1(RYR1)and purinergic receptor P2X and ligand-gated ion channel 3(P2RX3)(Yang et al.,2017).In addition to whole genome analysis in yak,Marco Polo sheep,and Tibetan antelope,resequencing analysis between low- and high-altitude ruminant populations has also identified candidate genes associated with hypoxia response, especially the HIF-1 pathway(Hu et al.,2019;Wang et al.,2016),including one gene(NOXA1)in goats(Wang et al.,2016)and eight genes(EPO, TLR4, PIK3CA, PRKCA, EGLN3, EGLN2, IFNGR2,and CUL2)with strong signals in sheep(Hu et al.,2019).Although candidate genes under positive selection can contribute to high-altitude adaptation,introgression may also play a crucial role. For instance, genes involved in the response-to-hypoxia pathway (e.g., EGLN1, EGLN2 and HIF3a)are introgressed from yak to Tibetan cattle,which may be responsible for their high-altitude adaptation(Wu et al.,2018).Of these genes,EGLN1 is reported to be associated with high-altitude adaptation in Tibetans (Simonson et al.,2010).Therefore,these positively selected genes may provide valuable insights into plateau medicine.
DOMESTICATION EFFECTS ON RUMINANT LIVESTOCK
The importance of ruminants is also highlighted by several major livestock species,including goat,sheep,cattle,and yak.Domestication of livestock ruminants was a key factor in the transition from hunter-gather societies to agricultural civilizations. Under intense artificial selection, domesticated livestock species have experienced extensive morphological or physiological changes compared to their wild relatives,including shadow color coat,floppy ears,shorter muzzles,smaller brain and cranial capacity,and tameness,which are referred to as domestication syndromes(Wilkins et al.,2014).
It has been hypothesized that domestication syndromes predominantly resulted from mild neural crest cell (NCC)deficits during embryonic development(Wilkins et al.,2014).Several recent studies have revealed that selected candidate regions under domestication of cattle,yak,sheep,and goat are enriched in pathways related to brain development or neurobehavioral functioning,such as NCCs(Alberto et al.,2018;Naval-Sanchez et al.,2018;Qanbari et al.,2014;Qiu et al., 2015). For example, neural genes (e.g., TMEM132D,CACNA1C, NRXN1, and NPAS3) show signals of positive selection in cattle(Qanbari et al.,2014).Thirty genes in yak show signals of selection involved in brain and neuronal development,19 of which are associated with behavior(Qiu et al.,2015).In addition,five genes are associated with the nerve system in sheep and goat,which showed convergent evolution(Alberto et al.,2018).These genes associated with the brain and nerve system under pressure from artificial selection may explain tameness or aggressiveness in livestock species.
In addition to major livestock species,reindeer were also domesticated by sub-Arctic people.Although cervid species are usually cautious and sensitive,reindeer are very tame and are the only domesticated species in the cervid family.Lin et al. (2019) identified reindeer-specific mutations in genes associated with NCCs to explain their tameness, and observed that genes involved in development(MSX2,ID3,BCAT1, CAD6, and CAD11), migration (TCOF1, BCAT1,NOTCH2,and NOTCH3),and differentiation(COL2A1,KIT,and SI)of NCCs were either rapidly evolving or demonstrated reindeer-specific mutations.
PERSPECTIVES
Traditional biological methods are often unable to solve the genetic basis of the evolution of complex traits across higher taxa at the family,order,or even class level.To address this issue, we propose a new approach called evolutionary genotype-phenotype systems biology (eGPS), which compares the evolution of many species using large-scale genome, transcriptome, regulatome, metabolome, and proteome data and systematically integrates evidence from genetic factors, developmental network evolution, and phenotypes to analyze the genetic basis of complex animal traits,especially those with evolutionary significance.Using this approach,we could resolve the genetic basis of complex traits and determine key regulatory genes or pathways.Functional experiments could verify these genetic factors,and thus facilitate further investigation on the evolutionary mechanisms of complex traits and their potential application.The Ruminant Genome Project has served as a good example of eGPS study with large-scale multi-omics data.
The rapid development of sequencing technology,especially long-read and Hi-C technologies, has enabled researchers to obtain high-quality reference genomes with long continuous contigs and scaffold size, even to the chromosome level.High-quality assemblies should promote additional studies on ruminants, including chromosome evolution,accurate identification of new functional elements(e.g., regulatory elements and new genes), and functional validation using gene editing technology such as CRISPR/Cas9. Chromosome numbers in Muntiacus species demonstrate considerable variations due to chromosome fusion;for example,Muntiacus reevesi has a karyotype of 2n=46,Muntiacus vaginalis has a karyotype of 2n=6 and 7,and Muntiacus crinifrons has a karyotype of 2n=8 and 9 in females and males,respectively(Shi,1983;Wurster&Benirschke,1967, 1970). High-quality reference genomes will help to reveal the genetic mechanisms underlying the recurrent fusion of chromosomes in muntjacs and its effects on the interaction between genes flanking the fusion regions,and thus facilitate understanding of speciation driven by chromosome variation.New regulatory elements and genes may be crucial in adaptive phenotypic diversification.Availability of high-quality genome data should facilitate identification of these new functional elements,thus revealing the potential genetic basis underlying adaptations. For casual or functional mutations identified using large-scale data among linages, CRISPR/Cas9 experiments could be utilized for verification using model organisms or key livestock species,such as sheep and goats.
Additionally,genomic resources and comparative analyses of ruminants are likely to contribute to animal husbandry(Figure 2).Comparative analysis between domestic livestock species and wild species has identified a series of candidate genes associated with domestication or economically valuable traits,such as meat quality and milk production(Alberto et al.,2018). These artificially selected genes can provide new targets for animal breeding and improvement.In the future,gene editing technology (such as CRISPR/Cas9) may be applicable to quickly improve traits of livestock.
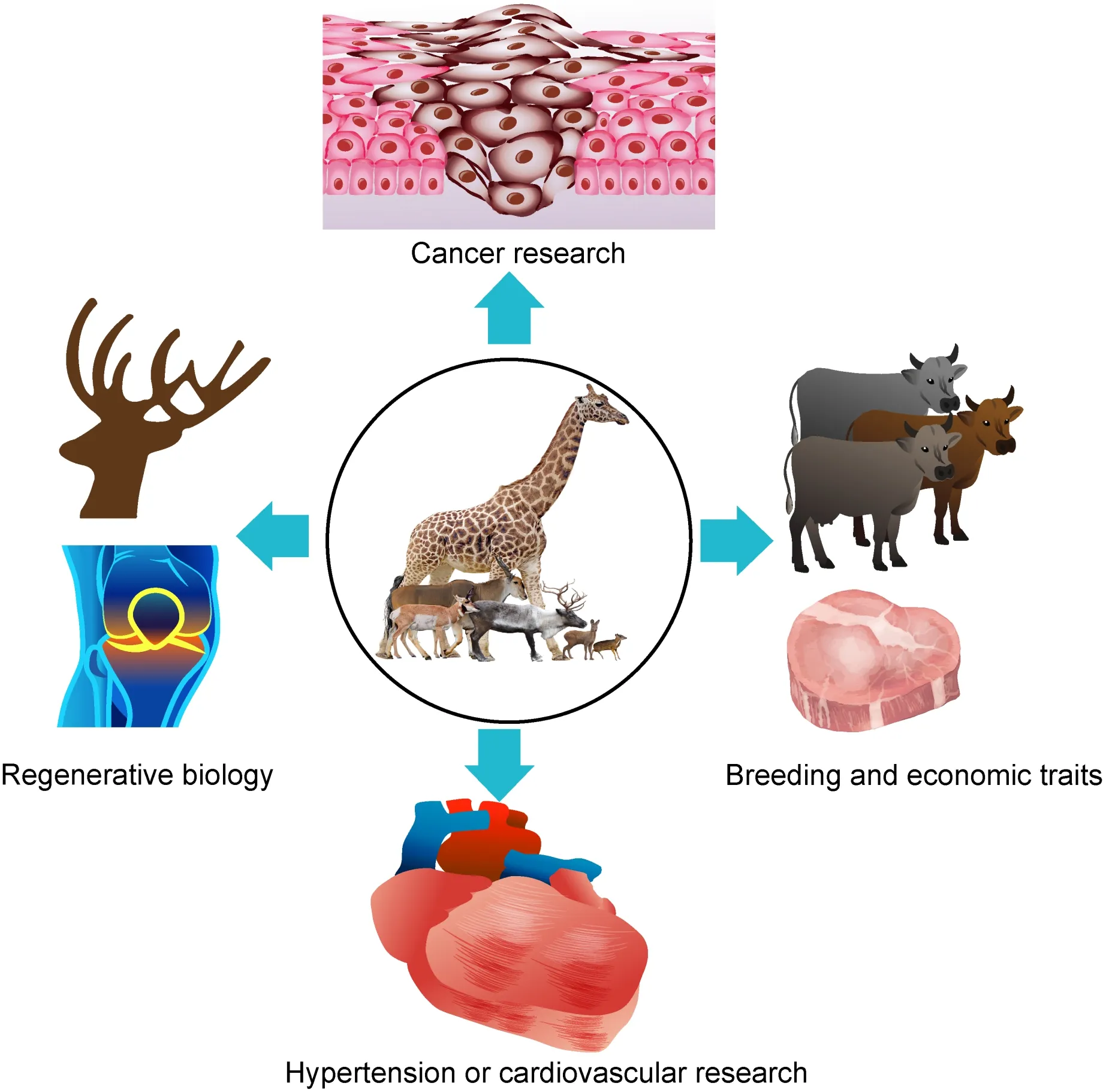
Figure 2 Possible applications of future ruminant research
In addition to application in agriculture, ruminants could serve as animal models for medical research and thus provide valuable insights into medical treatments and even clinical studies(Figure 2).Further research on deer antlers may offer unprecedented clues on regenerative biology and oncogenesis.Moreover,study on giraffes may provide insight into hypertension or cardiovascular research. Overall,increasing our understanding of the genetic basis and mechanisms underlying certain ruminant traits could benefit human society in many ways.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS'CONTRIBUTIONS
W.W.and L.C.proposed the ideas and revised the manuscript.B.W.collected data, reviewed the literature, and drafted the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We would like to thank three reviewers for their contributions to this work.
杂志排行

Zoological Research的其它文章
- From our roots,we grow Celebrating the 60th anniversary of the Kunming Institute of Zoology,Chinese Academy of Sciences
- Antimicrobial peptides:new hope in the war against multidrug resistance
- Chromosomal level assembly and population sequencing of the Chinese tree shrew genome
- Superior intestinal integrity and limited microbial translocation are associated with lower immune activation in SIVmac239-infected northern pig-tailed macaques(Macaca leonina)
- Conserved sequences identify the closest living relatives of primates
- Systematic relationships of Chinese freshwater semisulcospirids(Gastropoda,Cerithioidea)revealed by mitochondrial sequences