实时定量PCR在先天性小睑裂综合征患者FOXL2基因检测中的应用
2019-10-30陈丽娟王中英李思佳胡姗姗
陈丽娟 王中英 李思佳 胡姗姗
[摘要]目的 应用实时定量PCR技术鉴定先天性小睑裂综合征(BPES)患者中FOXL2基因的突变和(或)缺失。方法 于2017年8月收集黑龙江省1个BPES患病家系,采集外周血提取基因组DNA,用PCR直接测序法和实时定量PCR法筛查FOXL2基因的全部外显子。结果 在此例患病家系中,用PCR直接测序法排除了基因内突变,用实时定量PCR方法确定了FOXL2基因全缺失。这些变异在未患病亲属和50名正常人中并未被检测到。结论 本研究是应用实时定量PCR技术检测中国BPES患者FOXL2基因缺失的报道,这项技术丰富了BPES患者的分子遗传学诊断方法。
[关键词]小睑裂综合征;叉头蛋白L2;实时定量PCR;缺失;突变
[中图分类号] R777.1 [文献标识码] A [文章编号] 1674-4721(2019)8(b)-0079-04
[Abstract] Objective To identify the mutation or deletion of the FOXL2 gene in patients with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) using quantitative real-time PCR technology. Methods A family with BPES was collected in Heilongjiang Province in August 2017. Genomic DNA extracted from peripheral blood was collected from the family. PCR direct sequencing and quantitative real-time PCR sequencing for the whole exon of the FOXL2 gene were performed. Results In this family, deletion of the FOXL2 gene was confirmed by the quantitative real-time PCR technique, which intragenic mutations were excluded by PCR direct sequencing. This change was not detected either in the non-carrier relatives or in 50 normal controls. Conclusion This is the study to report FOXL2 gene deletion detected by quantitative real-time PCR in Chinese BPES patients. This technique enriches the diagnostic methods of molecular genetics in BPES patients.
[Key words] Blepharophimosis-ptosis-epicanthus inversus syndrome; Forkhead box protein L2; Quantitative real-time PCR; Deletion; Mutation
小瞼裂综合征(BPES)是一种罕见的遗传疾病,以眼睑畸形和卵巢功能不全为特征。依据是否伴有卵巢功能衰竭(POF),本病分为两种临床亚型,Ⅰ型患病女性同时患有POF;Ⅱ型则与POF无关[1-2]。BPES主要以常染色体显性方式遗传,偶有散发病例和常染色体隐性遗传方式报道[3]。经细胞重组和连锁分析研究后,BPES可能致病基因被定位于人类染色体3q23上[4]。随后,FOXL2(forkhead box protein L2)基因被确定为BPES的致病基因。此外,非综合征性POF和卵巢粒细胞瘤也相继报道与FOXL2的基因突变相关[5-6]。
迄今为止,已报道的与BPES患者相关的FOXL2基因突变有200余个。在所有已发现的遗传缺陷中,约有80%的病例是由于FOXL2基因内部突变所致[7];约2%的病例与细胞染色体的重排相关,包括染色体不平衡易位和3号染色体的中间缺失[8];约12%的病例是源于部分或全部的FOXL2基因长缺失和(或)其邻近的基因缺失[9];还有5%的病例是由于FOXL2的调控基因缺失[10]。目前有文献报道,可应用多重连接探针扩增(MPLA)技术[11]检测FOXL2基因单倍体缺失来证实常染色体显性遗传方式的BPES家系是源于FOXL2基因长缺失。本研究应用PCR直接测序法和实时定量PCR法对1个患有BPES中国家系进行FOXL2基因的突变及长缺失筛查。
1对象与方法
1.1实验对象
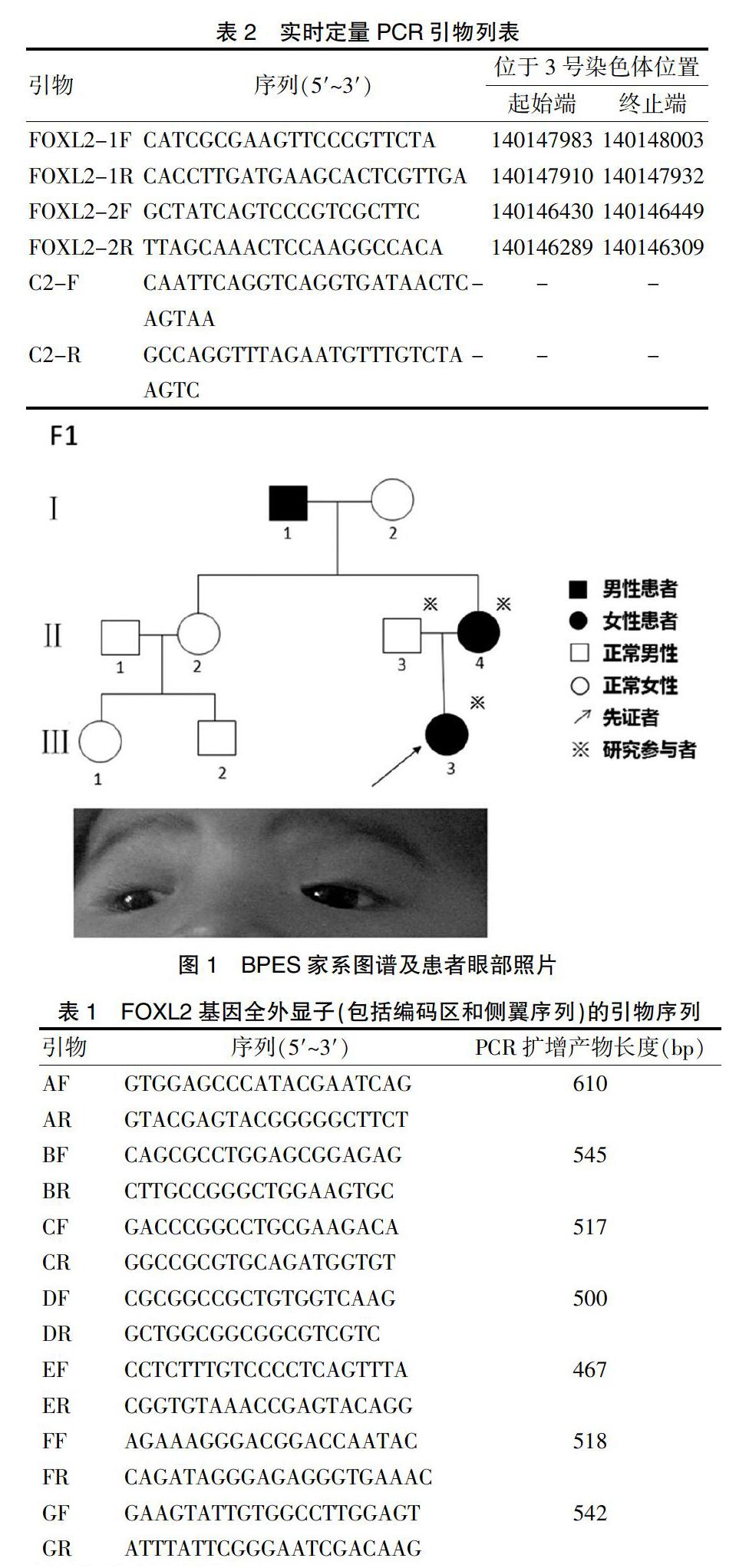
于2017年8月收集黑龙江省1个BPES患病家系(F1)。本研究经过牡丹江医学院医学伦理委员会批准,遵循赫尔辛基宣言,在知情同意的情况下,对家系中2位患者(先证者及其母亲)及1位有血缘关系的正常家系成员(先证者父亲)进行病史采集。对其家系患病情况进行系谱分析(3代9名成员,其中患有BPES者3名,未患病成员6名)。同时选取同期在我院体检中心进行健康检查的50名正常人,临床诊断由妇科和生殖科医师协诊,眼科医师确诊,排除其他眼部及系统疾病,且经由患者家属同意获取眼部照片。
1.2 PCR测序分析
血样采自BPES患者及正常家系成员,并且储存在-20℃的环境里。使用血液基因组DNA提取试剂盒(QIAamp DNA Mini kit,Qiagen,Hilden,Germany)抽提基因组DNA。应用聚合酶链反应(PCR)方法对FOXL2基因整个外显子(包括编码区和侧翼序列)DNA序列测定进行突变分析,引物序列如表1所示。
1.3实时定量PCR分析
实时定量PCR的引物由TakaRa生物工程公司设计、合成(引物序列如表2所示)。两对引物的扩增片段分别位于FOXL2基因的5′和3′端。实时定量PCR的反应体系按SYBR Green PCR Master Mix(TakaRa生物工程公司提供)试剂盒制备,应用7000实时定量PCR仪器进行实验操作。将位于21号染色体上的C2基因作为目标序列的量化模板。以正常对照组DNA作为校对标准,依据测得的相对循环周期阈值(Ct)来计算样本的相对拷贝数(RCN)[12]。实验均重复3遍,以排除人为误差。基因长缺失的判定点为A≈0.5倍RCN的异常。
2结果
2.1临床表现
本次研究中的先证者(F1-Ⅲ∶3,图1)为女童,有典型的BPES特征,即睑裂狭小、上睑下垂、倒转型内眦赘皮和内眦距离过宽。但POF需成人后确诊。其母亲患有BPES,在儿童期已接受过眼睑矫正术,目前未患POF。该家系未能确定BPES临床分型。
2.2 PCR测序结果
通过对FOXL2基因的直接测序,排除了家系中的基因内突变。
2.3实时定量PCR结果
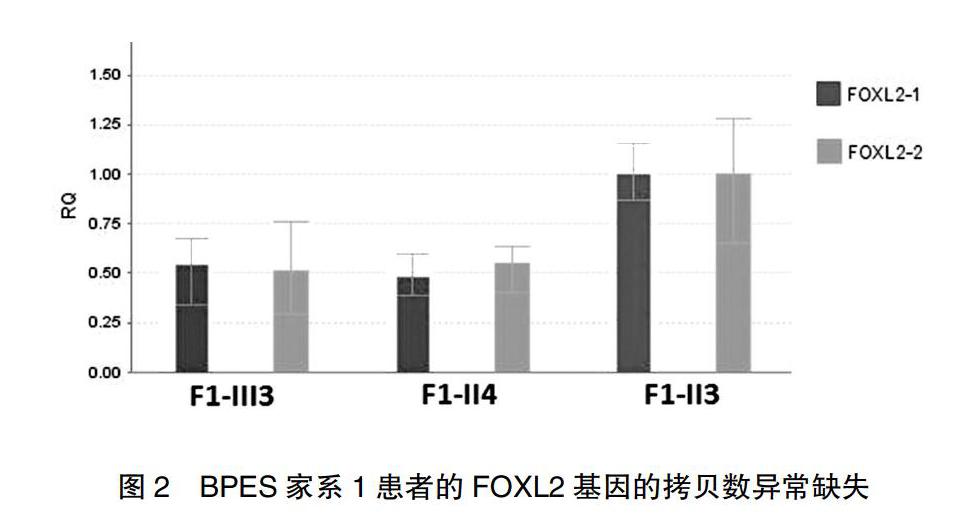
通过实时定量PCR技术进一步验证患者是否存在FOXL2基因的长缺失。研究显示,患者的RCN约为健康个体的一半(图2)。实时定量PCR检测到的产物拷贝数异常(CNVs)即意味着FOXL2基因的长缺失,缺失的长度为两组引物之间的全部FOXL2基因长度(chr 140148003-140146289)。实验均重复进行3次,以消除操作误差。正如实验设想所示,在BPES家系未患病亲属和50名正常人中均未检测到FOXL2基因的长缺失。实时定量PCR的两对外显子扩增产物表明家系患者的基因拷贝数约为未患病亲属的一半。
3讨论
本研究应用实时定量PCR技术在BPES家系(F1)中检测到了FOXL2基因的长缺失。与荧光原位杂交(FISH)技术和MLPA技术相比,实时定量PCR技术更为方便、快捷。本研究中分别位于FOXL2基因5′端和3′端的两对引物相对应的扩增片段的拷贝数均接近于健康个体的50%(图2),这就意味着这两对引物之间的区域缺失,即FOXL2基因长缺失。据报道,约12%的BPES病例是源于部分或全部的FOXL2基因长缺失和(或)其邻近的基因缺失,因而缺失筛查应常规应用于BPES的分子诊断领域。Beysen等[10]认为BPES患者FOXL2基因長缺失与POF的发生无相关性,而D′haene等[11]的研究表示,FOXL2的基因缺失可能与卵巢功能不全的严重程度相关。本研究试图在此家系中评估BPES的分型,家系1中患病女童处于青春期前的发展阶段,其母亲未表现出不孕或是患有POF,意味着FOXL2基因的长缺失未影响其在卵巢中的表达,所以BPES的分型不能被确定。FOXL2基因缺失引起的单倍剂量不足可能影响卵巢功能,导致女孩在以后的某时期患POF。因此,除了眼科的随诊,未确定的表型的年轻女性患者需要密切的内分泌和妇产科的随诊。本研究是通过实时定量PCR技术检测中国患者FOXL2基因缺失,所以可以确定实时定量PCR可以作为FOXL2的基因缺失筛查的可靠、方便、廉价的分子诊断工具,这能使得BPES患者的基因咨询工作更加便利,并且能够帮助扩展女性患者对于POF的临床随诊。
以往的研究显示,FOXL2基因内突变产生的截短蛋白质会大量的聚集在细胞核内[13],这些异常定位和聚集影响FOXL2核内定位,会严重损害它自身的DNA绑定功能,进而影响其与其它蛋白质的相互作用。研究显示,FOXL2突变后转录功能的降低不仅与蛋白的异常聚集有关,而且与氨基酸侧链的位置是否朝向螺旋结构的疏水核密切相关[14]。也有学者认为FOXL2突变受到上下游其他基因的协同控制而发挥作用,影响靶基因的调节[15],从而导致BPES发生。本研究中的FOXL2基因缺陷引起单倍体剂量不足,导致无效等位基因的产生,并可能导致转录物的失效,产生无意义的衰变[16]。由于FOXL2基因羧基末端的完整多聚丙氨酸长链对StAR基因(生成类固醇急性调控因子基因)的转录有抑制作用[17],因而FOXL2基因缺失或是失去多聚丙氨酸长链的基因突变可以增强StAR的表达,继而导致BPES和POF的发生,成为BPES的发病机制。
综上所述,本研究是通过实时定量PCR技术对中国人检测FOXL2基因缺失,为将实时定量PCR技术作为相对可靠的、便利的和廉价的方法应用于检测BPES患者遗传学缺陷提供理论支持。该技术的应用不仅促进了BPES的遗传学诊断发展,还为患病家系的遗传学咨询提供了依据。
[参考文献]
[1]Gulati R,Verdin H,Halanaik D,et al.Co-occurrence of congenital hydronephrosis and FOXL2-associated blepharophimosis,ptosis,epicanthus inversus syndrome (BPES)[J].Eur J Med Genet,2014,57(10):576-578.
[2]Nuovo S,Passeri M,Di Benedetto E,et al.Characterization of endocrine features and genotype-phenotypes correlations in blepharophimosis-ptosis-epicanthus inversus syndrome type 1[J].J Endocrinol Invest,2016,39(2):227-233.
[3]Chawla B,Bhadange Y,Dada R,et al.Clinical,radiologic,and genetic features in blepharophimosis,ptosis,epicanthus inversus syndrome in the Indian population[J].Invest Ophthalmol Vis Sci,2013,54(4):2985-2991.
[4]Crisponi L,Deiana M,Loi A,et al.The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome[J].Nat Genet,2001,27(2):159-166.
[5]Yang XW,He WB,Gong F,et al.Novel FOXL2 mutations cause blepharophimosis-ptosis-epicanthus inversus syndrome with premature ovarian insufficiency[J].Mol Genet Genomic Med,2018,6(2):261-267.
[6]Leung DT,Fuller PJ,Chu S.Impact of FOXL2 mutations on signaling in ovarian granulosa cell tumors[J].Int J Biochem Cell Biol,2016,72:51-54.
[7]Yang L,Li T,Xing Y.Identification of a novel FOXL2 mutation in a single family with both types of blepharophimosis-ptosis-epicanthus inversus syndrome[J].Mol Med Rep,2017, 16(4):5529-5532.
[8]Yang Y,Yang C,Zhu Y,et al.Intragenic and extragenic disruptions of FOXL2 mapped by whole genome low-coverage sequencing in two BPES families with chromosome reciprocal translocation[J].Genomics,2014,104(3):170-176.
[9]Bouman A,van Haelst M,van Spaendonk R.Blepharophimosis-ptosis-epicanthus inversus syndrome caused by a 54-kb microdeletion in a FOXL2 cis-regulatory element[J].Clin Dysmorphol,2018,27(2):58-62.
[10]Beysen D,Raes J,Leroy BP,et al.Deletion involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome[J].Am J Hum Genet,2005, 77(2):205-218.
[11]D′haene B,Nevado J,Pugeat M,et al.FOXL2 copy number changes in the molecular pathogenesis of BPES:unique cohort of 17 deletions[J].Hum Mutat,2010,31(5):E1332-E1347.
[12]Sun M,Ma F,Zeng X,et al.Triphalangeal thumb-polysyndactyly syndrome and syndactyly type Ⅳ are caused by genomic duplications involving the long range,limb-specific SHH enhancer[J].J Med Genet,2008,45(9):589-595.
[13]Krepelova A,Simandlova M,Vlckova M,et al.Analysis of FOXL2 detects three novel mutations and an atypical phenotype of blepharophimosis-ptosis-epicanthus inversus syndrome[J].Clin Exp Ophthalmol,2016,44(9):757-762.
[14]Beysen D,Moumne L,Veitia R,et al.Missense mutations in the forkhead domain of FOXL2 lead to subcellar mislocalization,protein aggregation and impaired transactivation[J].Hum Mol Genet,2008,17(13):2030-2038.
[15]Kim JH,Bae J.Differential apoptotic and proliferative activities of wild-type FOXL2 and blepharophimosis-ptosis-epicanthus inversus syndrome (BPES)-associated mutant FOXL2 protein[J].J Reprod Dev,2014,60(1):14-20.
[16]程洪波,王濤,王改改,等.一个睑裂狭小综合征家系FOXL2基因的突变分析[J].中华医学遗传学杂志,2018, 35(4):515-517.
[17]Pisarska MD,Bae J,Klein C,et al.Forkhead l2 is expressed in the ovary and represses the promoter activity of the steroidogenic acute regulatory gene[J].Endocrinology,2004,145(7):3424-3433.
(收稿日期:2019-02-18 本文编辑:祁海文)
