福白菊化学成分LC-MSn定性分析及HPLC-UV法多成分含量测定
2019-10-30李君彦范氏英孙中宣程雪翔叶文才
李君彦,范氏英,孙中宣,程雪翔,叶文才,汪 豪*
(1中国药科大学中药制药系,南京 210009;2湖北凤凰白云山药业有限公司,麻城 438300;3暨南大学药学院中药及天然药物研究所,广州 510632)
菊花为菊科植物菊(ChrysanthemummorifoliumRamat.)的干燥头状花序。味甘、苦,微寒,具有散风清热、平肝明目、清热解毒之功效,可用于目赤肿痛、眼目昏花、风热感冒、头痛眩晕等症。菊花药材按照产地和加工方法的不同,分为“亳菊”“滁菊”“贡菊”“杭菊”和“怀菊”等5种[1]。福白菊(Chrysanthemummorifolium‘Fubaiju’)主要产自我国湖北省麻城市,种植规模3万亩以上,是我国三大主要白菊品种之一,为湖北省重要道地药材[2]。菊花主要化学成分为黄酮、挥发油、有机酸、多糖和氨基酸等。药理研究表明,菊花中黄酮类化合物具有调血脂、抗肿瘤、免疫调节等生物活性,酚酸类化合物具有抑菌等药效活性[3]。目前,福白菊化学成分及质量研究文献报道较少,曾报道福白菊挥发油成分的定性分析[4]。《中华人民共和国药典》中菊花质量标准收载绿原酸、木犀草苷、3,5-二咖啡酰基奎宁酸等3个指标成分含量测定项[1]。因此,本文拟采用LC-MSn技术对福白菊醇提物化学成分进行定性分析;并采用HPLC-UV法,同一色谱条件下测定福白菊中5种化学成分含量,包括:绿原酸(1)、木犀草素-7-O-β-D-葡萄糖苷(2)、木犀草素-7-O-β-D-葡萄糖醛酸苷(3)、3,5-二咖啡酰基奎宁酸(4)、芹菜素-7-O-β-D-葡萄糖苷(5)(结构式见图1),以期快速、准确地定性、定量分析福白菊中的主要化学成分,为福白菊的药效物质基础及质量控制研究奠定研究基础。
Figure1 Structures of the five major constituents fromChrysanthemummorifolium‘Fubaiju’
1 材 料
1.1 药材与试剂
福白菊药材主要采集于湖北省麻城市等地,产地及批号见表1,经中国药科大学秦民坚教授鉴定为福白菊(Chrysanthemummorifolium‘Fubaiju’)。实验用标准品绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷、4,5-二咖啡酰基奎宁酸均来源于中国食品药品检定研究院,纯度大于98.0%,结构式见图1。HPLC用乙腈和甲醇为色谱纯;其他试剂均为分析纯。
Table1 Origin and batch number ofChrysanthemummorifolium‘Fubaiju’

SampleNo.OriginBatchNo.1Macheng,Hubeiprovince1611252Macheng,Hubeiprovince1710023Macheng,Hubeiprovince1605024Macheng,Hubeiprovince161125B5Macheng,Hubeiprovince161125B26Xianyangyan,Hubeiprovince181106A27Xianyangyan,Hubeiprovince181107A38Xianyangyan,Hubeiprovince181105A49Linjiawan,Hubeiprovince181108B310Xianyangyan,Hubeiprovince181119A4
1.2 仪 器
液相色谱仪(含LC-20AT四元泵、SIL-20A HT自动进样器、CTO-20AC柱温箱、SPD-M20A型检测器),AUW120D型电子天平(日本岛津公司);6520 Accurate-Mass Q-TOF液质仪,1200高效液相色谱仪,MassHunter B.04.00定性分析软件(美国安捷伦公司)。
2 方法与结果
2.1 LC-MS定性分析
2.1.1 样品制备
对照品溶液的制备 分别取绿原酸、木犀草素-7-O-葡萄糖苷、木犀草素-7-O-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-葡萄糖苷和4,5-二咖啡酰基奎宁酸对照品适量,精密称定,加70%甲醇溶解,制成混合对照品溶液,滤过,即得。
供试品溶液的制备 取福白菊粉末(过一号筛,批号:161125)约25 mg,精密称定,加 50%甲醇10 mL,超声提取60 min,0.45 μm微孔滤膜滤过,作为供试品溶液。
2.1.2 HPLC条件 色谱柱:Shim-pack VP-ODS(150 mm×2.0 mm,5 μm),柱温30 ℃。流动相A:0.1%甲酸溶液,流动相B:乙腈,梯度洗脱:0~10 min,B:15%~22%;10~15 min,B:22%~23%;15~20 min,B:23%;20~35 min,B:23%~30%。流速:0.3 mL /min;进样量:10 μL。
2.1.3 MS条件 离子源为ESI,负离子模式,雾化气为氮气。去溶剂气体温度:325 ℃,去溶剂气体流速:8 mL/min,喷雾器压力:40 psi(1 psi=6.895 kPa),毛细管电压:4 000 V,碎裂电压:100 V,碰撞能量:30 V,质谱检测范围:MS:m/z100~1 000;MS/MS:m/z100~800。
2.1.4 实验结果 通过“2.1.2”项下色谱条件对福白菊样品溶液进行MS分析,负离子模式下总离子流图见图2。有标准品的化合物通过与标准品的色谱保留时间以及质谱裂解碎片进行确认;没有标准品的未知成分,根据色谱保留时间、质谱裂解碎片、紫外吸收并结合文献进行确认,共鉴定了22个化合物,见表2。
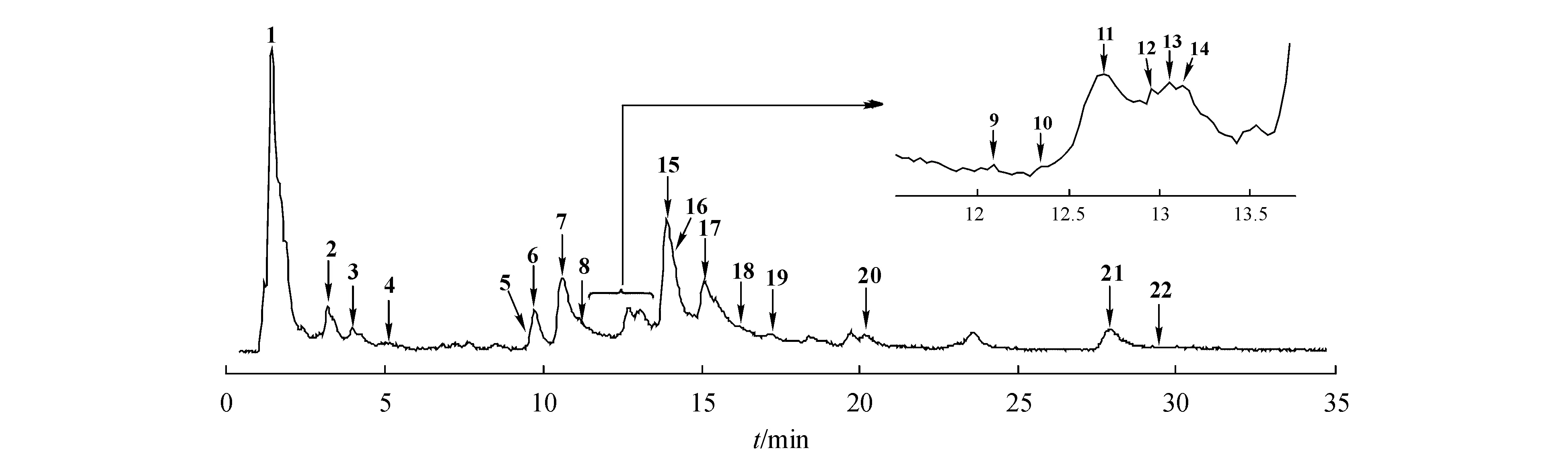
Figure2 Negative total ion chromatogram ofChrysanthemummorifolium‘Fubaiju’
Table2Identification of chemical consitituents ofChrysanthemummorifolium‘Fubaiju’
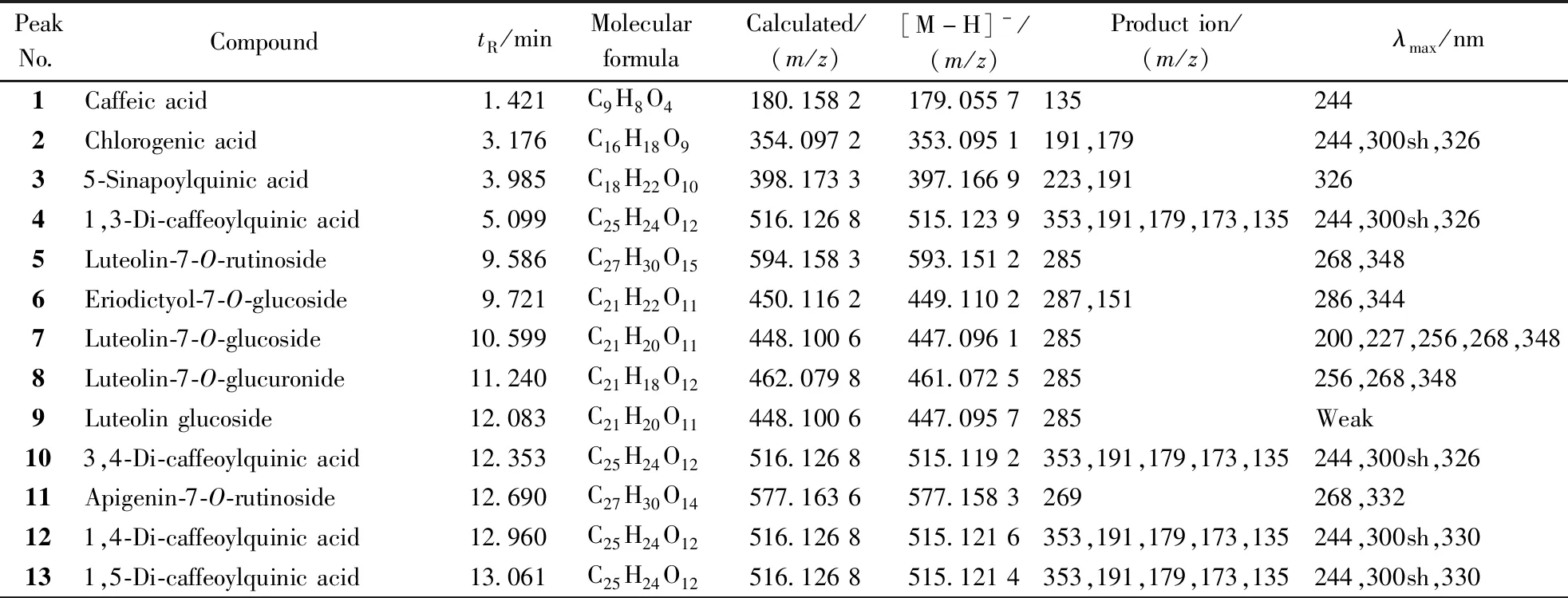
PeakNo.CompoundtR/minMolecular formula Calculated/(m/z)[M-H]-/(m/z)Production/(m/z)λmax/nm1Caffeicacid1.421C9H8O4180.1582179.05571352442Chlorogenicacid3.176C16H18O9354.0972353.0951191,179244,300sh,32635-Sinapoylquinicacid3.985C18H22O10398.1733397.1669223,19132641,3-Di-caffeoylquinicacid5.099C25H24O12516.1268515.1239353,191,179,173,135244,300sh,3265Luteolin-7-O-rutinoside9.586C27H30O15594.1583593.1512285268,3486Eriodictyol-7-O-glucoside9.721C21H22O11450.1162449.1102287,151286,3447Luteolin-7-O-glucoside10.599C21H20O11448.1006447.0961285200,227,256,268,3488Luteolin-7-O-glucuronide11.240C21H18O12462.0798461.0725285256,268,3489Luteolinglucoside12.083C21H20O11448.1006447.0957285Weak103,4-Di-caffeoylquinicacid12.353C25H24O12516.1268515.1192353,191,179,173,135244,300sh,32611Apigenin-7-O-rutinoside12.690C27H30O14577.1636577.1583269268,332121,4-Di-caffeoylquinicacid12.960C25H24O12516.1268515.1216353,191,179,173,135244,300sh,330131,5-Di-caffeoylquinicacid13.061C25H24O12516.1268515.1214353,191,179,173,135244,300sh,330
(Continued)

PeakNo.CompoundtR/minMolecular formula Calculated/(m/z)[M-H]-/(m/z)Production/(m/z)λmax/nm143,5-Di-caffeoylquinicacid13.131C25H24O12516.1268515.1213353,191,179,173,135244,300sh,33015Apigenin-7-O-glucoside13.905C21H20O10432.1056431.1003269268,33216Diosmetin-7-O-rutinoside14.074C28H32O15608.5582607.1691299254,268,348174,5-Di-caffeoylquinicacid15.052C25H24O12516.1297515.1225353,191,179,173,135244,300sh,33018Diosmetin-7-O-glucuronide16.267C22H20O12476.0955475.0898299254,268,34819Apigenin-7-O-6″-malonylglucoside17.144C24H22O13518.4200517.1906269Weak20Luteolin-7-O-6″-acetylglucoside20.147C23H22O12490.1111489.1062285Weak21Apigenin-7-O-6″-acetylglucoside27.941C23H22O11474.1162473.1101269Weak22Acacetin-7-O-glucuronide29.763C22H20O11460.1006459.0954283Weak
峰7、8、15与混合对照品质谱图进行比对,分别为luteolin-7-O-glucoside、luteolin-7-O-glucuronide、apigenin-7-O-glucoside。
峰5、峰9和峰20与峰7具有相似的裂解过程,均产生m/z285的碎片离子,推断其均为luteolin类衍生物。根据其准分子离子峰,结合保留时间和紫外最大吸收波长及与文献[5]对比,峰9与峰7具有相同的分子离子峰m/z447[M-H]-和碎片m/z285[M-H-Glucose]-,但其保留时间约滞后1.5 min,故推测其为luteolin glucoside,糖基连接位置无法确定;峰5具有m/z593[M-H]-,比luteolin-7-O-glucoside的分子离子峰大92,故推断其为luteolin-7-O-rutinoside;峰20的分子离子峰m/z489[M-H]-比luteolin-7-O-glucoside的分子离子峰大42,推断其为luteolin-7-O-glucoside的乙酰基衍生物,故鉴定为luteolin-7-O-6″-acetylglucoside。
峰6在ESI-模式下得到m/z449[M-H]-,其二级质谱得到m/z287[M-H-Glucose]-,且在286和344 nm处有最大吸收,结合保留时间及与文献[5]对比,鉴定其为eriodictyol-7-O-glucoside。
峰11、峰19和峰21与峰15具有相似的裂解途径,二级质谱均得到m/z269的碎片离子。峰11在ESI-模式下得到m/z577[M-H]-,比apigenin-7-O-glucoside的分子离子峰大92,其二级质谱得到m/z269[M-H-Glucose]-,且在268 nm和332 nm处有最大吸收,结合保留时间及与文献[5]对比,鉴定其为apigenin-7-O-rutinoside;峰19的分子离子峰m/z517[M-H]-比apigenin-7-O-glucoside的分子离子峰大86,推断其为apigenin-7-O-glucoside的丙二酸单酰基衍生物,结合其保留时间及与文献[6]对比,故鉴定为apigenin-7-O-6″-malonylglucoside;峰21的分子离子峰m/z473[M-H]-比apigenin-7-O-glucoside的分子离子峰大42,结合其保留时间及与文献[5]对比,推断其为apigenin-7-O-glucoside的乙酰基衍生物,故鉴定为apigenin-7-O-6″-acetylglucoside。
峰16在ESI-模式下可得到m/z607[M-H]-,二级质谱得到m/z299[M-H-Rutinose]-,结合保留时间和紫外最大吸收波长及与文献[5]对比,推断其为diosmetin-7-O-rutinoside。峰18在ESI-模式下可得到m/z475[M-H]-,二级质谱得到m/z 299[M-H-Glucuronide]-,结合保留时间和紫外最大吸收波长及与文献[5]对比,推断其为diosmetin-7-O-glucuronide。
峰22在ESI-模式下可得到m/z459[M-H]-,二级质谱得到m/z283[M-H-Glucuronide]-,结合保留时间和紫外最大吸收波长及与文献[5]对比,推断其为acacetin-7-O-glucuronide。
峰2、峰14、峰17峰与混合对照品质谱图进行比对,分别为chlorogenic acid、3,5-di-caffeoylquinic acid、4,5-di-caffeoylquinic acid。
峰1在ESI-模式下得到m/z179[M-H]-,其二级质谱得到m/z135[M-H-CO2]-,且在244 nm处有最大吸收,结合保留时间及与文献[5]对比,鉴定其为caffeic acid。
峰3在ESI-模式下得到m/z 397[M-H]-,具有碎片离子m/z223[sinapoyl-H]-,且在326 nm 处有最大吸收,结合保留时间及与文献[5,7]对比,鉴定其为5-sinapoylquinic acid。
峰4、峰10、峰12和峰13在ESI-模式下均得到m/z515[M-H]-,且均可得到多级碎片m/z353[M-H-Caffeoyl]-、m/z191[M-H-2Caffeoyl]-、m/z179[Caffeic acid-H]-、m/z173[Quinic acid-H-H2O]-、m/z135[Caffeic acid-H-CO2]-,根据化合物出峰顺序不同,结合文献[5-6,8],推断峰4为1,3-di-caffeoylquinic acid,峰10为3,4-di-caffeoylquinic acid,峰12为1,4-di-caffeoylquinic acid,峰13为1,5-di-caffeoylquinic acid。
2.2 HPLC-UV含量测定
2.2.1 HPLC-UV溶液的制备
对照品溶液的制备 分别取绿原酸(1)、木犀草素-7-O-β-D-葡萄糖苷(2)、木犀草素-7-O-β-D-葡萄糖醛酸苷(3)、3,5-二咖啡酰基奎宁酸(4)、芹菜素-7-O-β-D-葡萄糖苷(5)对照品适量,精密称定,加70%甲醇制成每毫升含绿原酸30.0 μg、木犀草素-7-O-β-D-葡萄糖苷39.6 μg、木犀草素-7-O-β-D-葡萄糖醛酸苷29.8 μg、3,5-二咖啡酰基奎宁酸60.4 μg、芹菜素-7-O-β-D-葡萄糖苷50.0 μg的混合对照品溶液,滤过,即得。
供试品溶液的制备 取福白菊药材粉末(过一号筛)约0.25 g,精密称定,精密加入70%甲醇25 mL,称定重量,超声提取40 min,放冷,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.2.2 色谱条件及系统适用性
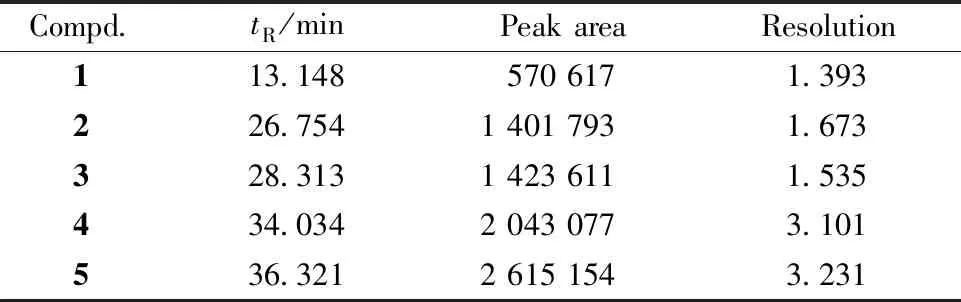
Shim-pack VP-ODS(250 mm×4.6 mm,5 μm)色谱柱,流动相A:0.1%磷酸溶液,流动相B:乙腈,梯度洗脱:0 min~11 min,B:10%~18%;11 min~30 min,B:18%~20%;30 min~40 min,B:20%。流速为1.0 mL/min,检测波长为348 nm,柱温为30 ℃,进样量为10 μL。理论塔板数以3,5-二咖啡酰基奎宁酸峰计算不低于8 000,且5种化合物均能达到基线分离。供试品溶液及混合对照品溶液HPLC色谱图见图3,供试品溶液中5种化合物分离度如表3所示。
2.2.3 方法学考察
线性关系 分别吸取混合对照品溶液1.0,2.0,4.0,6.0,8.0,10.0,20.0,40.0 μL,注入液相色谱仪,记录峰面积。以峰面积(Y)为纵坐标,进样量(X,μg)为横坐标,进行线性回归。结果如表4所示,5种对照品在对应线性范围内相关系数(r)均大于0.999 9,呈良好线性。
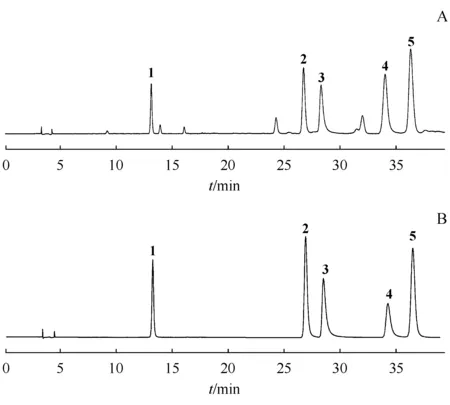
Figure3 (A) HPLC chromatogram ofChrysanthemummorifolium‘Fubaiju’ (batch No.161125);(B) HPLC chromatogram of reference substances
1:Chlorogenic acid;2:Luteolin-7-O-β-D-glucopyranoside;3:Luteolin-7-O-β-D-glucopyranuronide;4:3,5-Di-caffeoylquinic acid;5:Apigenin-7-O-β-D-glucopyranoside
Table3 Resolution of the five major constituents fromChrysanthemummorifolium‘Fubaiju’
Compd.tR/minPeakareaResolution113.1485706171.393226.75414017931.673328.31314236111.535434.03420430773.101536.32126151543.231
Table4 Calibration curves of the five major constituents fromChrysanthemummorifolium‘Fubaiju’

Compd.Linearrange/μgLinearequationrLOQ/ngLOD/ng10.03000-1.200Y=51624X+134680.99992.40.720.03960-1.584Y=116169X-219941.0003.21.630.02980-1.192Y=88790X-554180.99999.52.440.06040-2.416Y=132066X-293271.0009.74.350.05000-2.000Y=148256X-265911.0004.01.2
定量限(LOQ)和检测限(LOD) 取以上混合对照品溶液,用70%甲醇逐步稀释后进样分析,取(S/N≥10)和(S/N≥3)信噪比时的进样量分别为定量限和检测限。结果如表4 所示,绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷的定量限(S/N≥10)分别为2.4、3.2、9.5、9.7、4.0 ng,检测限(S/N≥3)分别为0.7、1.6、2.4、4.3、1.2 ng。
精密度 精密吸取混合对照品溶液,连续进样6次,记录峰面积。结果绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷的峰面积RSD分别为0.11%、0.20%、1.00%、1.71%、1.70%,表明本法精密度较好。
重复性 取同一批福白菊(批号:161125)粉末6份,按“2.2.1”项下方法平行制备6份供试品溶液,按照上述色谱条件检测。结果显示,绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷的含量(%)分别为0.34、0.64、0.48、1.04和1.23,RSD(%)分别为0.72、0.19、1.49、0.85和0.77。结果表明,本法重复性较好。
稳定性 精密量取同一供试品溶液(批号:161125)10 μL,4 ℃保存,分别于0、2、4、8、12、24 h进样,记录绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷的峰面积。结果显示,上述5种成分峰面积的RSD(%,n=6)分别为0.93、0.36、0.52、0.75和1.36,表明供试品溶液在4℃放置24 h内基本稳定。
加样回收率 取已准确测定含量的福白菊(批号:161125)粉末0.25 g,共9份,精密称定,分别加入高、中、低浓度的对照品适量,每个浓度设置3份,按“2.2.1”项下方法制备供试品溶液,进样检测,按外标法计算绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷的加样回收率和RSD。结果显示,5种成分的加样回收率(%)分别为103.16、100.99、101.14、99.41和98.72,RSD(%)分别为0.66、0.79、1.07、1.31和1.18,均符合规定。
2.2.4 样品测定 收集10批福白菊药材(产地、批号等信息见表1),按“2.2.1”项下方法制备供试品溶液,在上述色谱条件下进行样品测定,按外标法计算样品中绿原酸(1)、木犀草素-7-O-β-D-葡萄糖苷(2)、木犀草素-7-O-β-D-葡萄糖醛酸苷(3)、3,5-二咖啡酰基奎宁酸(4)、芹菜素-7-O-β-D-葡萄糖苷(5)含量。结果见表5。
Table5 Contents determination ofChrysanthemummorifolium‘Fubaiju’ samples

BatchNo.Content/%123451611250.320.630.460.971.231710020.480.440.651.270.511605020.300.410.520.880.68161125B0.290.480.410.820.81161125B20.300.440.450.890.80181106A20.500.550.561.150.62181107A30.480.530.521.100.59181105A40.510.530.591.190.58181108B30.650.510.651.340.60181119A40.320.260.300.700.29
1:Chlorogenic acid;2:Luteolin-7-O-β-D-glucopyranoside;3:Luteolin-7-O-β-D-glucopyranuronide;4:3,5-Di-caffeoylquinic acid;5:Apigenin-7-O-β-D-glucopyranoside
3 讨 论
本研究采用LC-MSn法进行福白菊主要化学成分定性分析,建立HPLC-UV法同一色谱条件下5个指标成分含量测定方法并进行方法学研究。实验分别考察了检测波长、柱温和流动相种类,化合物1和4的最大吸收波长为327 nm,化合物2、3和5的最大吸收波长约在348 nm,参考《中华人民共和国药典》菊花药材含量测定的色谱条件,确定检测波长为348 nm;LC-MSn法进行定性分析时,流动相加入甲酸可改善色谱峰拖尾现象,离子化信息较为丰富;HPLC-UV法进行主要化合物的含量测定时,流动相选择为乙腈-0.1%磷酸水溶液,各成分分离效果良好。
本实验为首次对福白菊醇提物的化学成分进行LC-MSn定性分析,通过对照品保留时间、碎片离子比对,及未知色谱峰的精确分子质量、裂解碎片等质谱信息分析,共鉴定出22个化学成分,包括13个黄酮类成分和9个酚酸类成分。采用HPLC-UV法同时测定福白菊中绿原酸、木犀草素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖醛酸苷、3,5-二咖啡酰基奎宁酸和芹菜素-7-O-β-D-葡萄糖苷5种主要成分的含量,10批福白菊药材中上述5种成分的含量分别为0.29%~0.65%、0.26%~0.63%、0.30%~0.65%、0.70%~1.34%、0.29%~1.23%,均符合《中华人民共和国药典》菊花品种的含量要求,其中3,5-二咖啡酰基奎宁酸,及芹菜素-7-O-β-D-葡萄糖苷为福白菊药材中含量较高成分。在不同采摘时间和不同产地的药材中,各成分含量不尽相同,药材1~9均为胎菊,药材10为朵菊,药材10各成分含量明显偏低,推测福白菊胎菊有效成分含量较高,品质优于朵菊。
