超临界流体萃取与模拟移动床色谱纯化灵芝三萜类化合物
2019-10-29余书奇包晓青梁明在金晨钟
余书奇,包晓青,梁明在*,金晨钟,田 蔚
(1.湖南人文科技学院农业与生物技术学院,湖南 娄底 417000;2.璞励卓越(北京)科技有限公司,北京 100000;3.义守大学化工系,中国台湾 高雄 84001)
灵芝作为传统的中药材具有很高的药用价值,已经成为保健食品的主要原料之一。研究发现灵芝具有调节免疫系统、心血管系统等药理功能[1],其中灵芝三萜类化合物是灵芝的关键药效成分之一,主要指标成分为灵芝酸A、灵芝酸F和灵芝醇B,现代药理学研究表明灵芝三萜类化合物具有抗肿瘤、保肝护肝、抗菌消炎等功能[2-6]。因此,对灵芝中的灵芝三萜类化合物进行有效提取及纯化,可拓宽灵芝产品多元化发展。目前,灵芝三萜类化合物的提取多采用有机溶剂提取、超声波辅助提取等方式。而采用超临界二氧化碳提取可提高灵芝三萜类化合物的稳定性,并且绿色无污染,安全无毒[7-9]。研究显示超临界流体萃取的操作条件对产物中灵芝三萜类化合物的组分种类和活性有一定程度的影响[10-13],因此本实验优化了超临界流体萃取的最佳条件与方法;同时采用模拟移动床(simulated moving bed,SMB)移除粗萃物中的不纯物以提高灵芝三萜类化合物含量。SMB技术是一种现代化精细的色谱分离技术,在固定床和真实移动床的基础上发展而来[14-15]。该技术采用固定相与移动相连续式地逆向流动,大幅提高了固定相的使用效率,达到连续进料提高产量的目的[15-17]。由于其相对于传统制备色谱分离技术具有能连续化操作、易实现自动化、分离能力强、分离效率高等特点,因而在食品科学和生物化学、医学领域得到了越来越广泛的应用[18-22],目前,食品领域中SMB的应用热点在天然产物的提取纯化上,如铁皮石斛[23]、白藜芦醇[24]、紫杉醇[25]、甜叶菊苷[26]、甘草苷[27]等的纯化,曾有研究者利用使用超临界流体的SMB进行了中国台湾牛樟芝三萜的分离纯化研究[28],但国内外尚未有SMB纯化灵芝三萜类化合物的应用报道。
1 材料与方法
1.1 材料与试剂
灵芝子实体由浙江寿仙谷公司提供,为赤灵芝。
灵芝酸A(>98%)、灵芝酸F(>98%)、灵芝醇B(>98%)标准品 成都曼思特股份有限公司;95%乙醇景明化工股份有限公司;乙腈(色谱纯) 友和贸易股份有限公司;二氧化碳30 kg(插管、高纯度)锦德气体股份有限公司。
1.2 仪器与设备
高效液相色谱( high performance liquid chromatography,HPLC,泵:2130,紫外检测器:L-2455)仪 日本Hitachi公司;超临界萃取设备(萃取槽体积为1 L,10.5 cm×11.1 cm) 中国台湾金属工业研究中心;SMB色谱(1/8不锈钢配管,配置8 支管柱,1.0 cm×25 cm) 中国台湾乔璞科技有限公司。
1.3 方法
1.3.1 超临界流体萃取条件优化
取1 3 7 g的灵芝子实体切粒(颗粒直径约1~3 mm),置于萃取槽中,在350 bar、45 ℃进行萃取,每0.5 h从分离槽中取样一次,分离槽温度设定为50 ℃,压力设定为45 bar,持续萃取6 h,根据萃取液中目标物含量的变化确定萃取时间。考察夹带剂对萃取结果的影响:A组不添加夹带剂,二氧化碳流速为60 g/min,在萃取过程中同时从萃取槽出口端以5 mL/min流速泵入乙醇溶液,以避免萃取物阻塞;B组采用乙醇为夹带剂,流速为5 mL/min,二氧化碳流速为60 g/min,在进入萃取槽前便与二氧化碳混合后再加热,共同进入萃取槽中萃取。
1.3.2 萃取液中灵芝三萜类化合物含量的测定
采用1.3.1节B组的方式对灵芝进行萃取,所得萃取液用HPLC测定目标组分含量。分析方法参照文献[29]:色谱柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm,5 μm),流动相按表1进行梯度洗脱,分析波长252 nm,流速1 mL/min,进样量20 μL。绘制灵芝酸A、灵芝醇B及灵芝酸F标准曲线,所得曲线相关系数均在0.999 5以上,线性范围在0~500 mg/L,通过内标法测定灵芝三萜类化合物的含量。

表1 流动相梯度洗脱方法Table 1 Mobile phase gradient elution program
3 种标准品的HPLC如图1所示,灵芝酸A的保留时间为35.1 min,灵芝酸F的保留时间为53.0 min,灵芝醇B的保留时间为86.5 min。
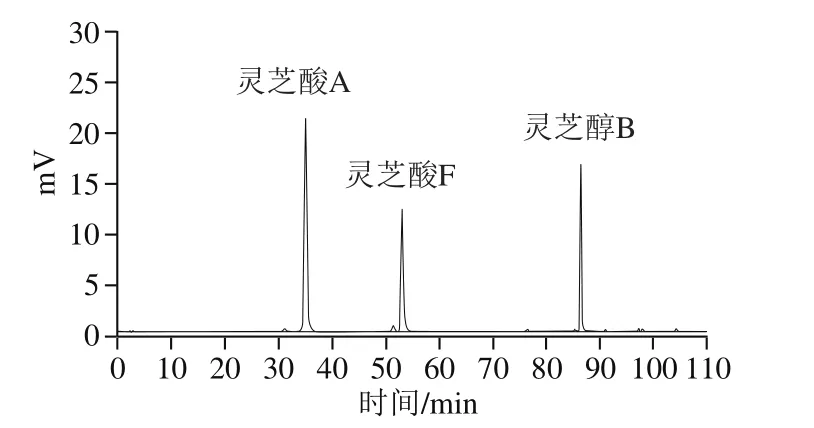
图1 灵芝三萜类化合物标准品液相色谱图Fig. 1 Liquid chromatogram of triterpenoid standards
1.3.3 SMB初始参数确定
按1.3.1节B组的方法萃取灵芝,所得萃取液作为SMB的进样原料。选取SMB系统8 只管柱中的1只管柱作为HPLC的分析柱,将C18填料填充于1.0 cm×25 cm的不锈钢管柱中。采用95%乙醇溶液及0.01%盐酸溶液作为流动相。考察不同流动相配比(乙醇溶液与盐酸溶液体积比为95∶5、90∶10、85∶15、50∶50、45∶55、40∶60)对分离结果的影响,从而选择适合SMB系统的流动相。
1.3.4 SMB参数优化设计
以1.3.1节B组方法所得到的萃取液为进样原料,以C18为固定相,选择1.3.3节最佳比例的乙醇-盐酸溶液作为流动相。通过优化SMB操作参数:不同的管柱设计、流动相配比及多通阀的切换时间等分离纯化灵芝三萜类化合物。分离实验共分两组进行:第1组目的是将低极性杂质分离,第2组目的是将高极性杂质分离。
1.3.5 三角形理论
SMB参数设计主要包括各出入口端流速的设定与多通阀的切换时间,本研究SMB参数的设定以三角形理论为基础[30-32],“三角形理论”法是在线性条件或非线性条件下,对操作SMB快速选择合适实验条件的有用工具。它能用于确定最佳条件,并可通过公式在高产率和短切换时间上得到一个折中方案。其主要参数mj为第j区段移动相的体积流速与固体体积流速的比值,定义如下:

式中:Qj为j区段流体的体积流速;VC为空管柱体积(本研究所采用的管柱体积为11.78 mL);tsw为所设定的多通阀切换时间周期;εt为管柱的孔隙度,即柱横截面上流动相所占的分数,为管柱内流动相体积与柱总体积之比;Vd为死体积,即管柱中未被固定相占据的空隙体积。
本研究进一步假设εt=0.35,并忽略死角体积Vd=0。
1.4 数据统计及图表绘制
SMB结果数据统计方法:SMB系统平衡稳定后,收取一个循环周期每个端口的样品,旋转蒸发挥干溶剂后,定容,通过液相色谱定量分析目标组分的浓度。
三角形绘制方法:通过式(2)[33-34]计算SMB中各组分的亨利常数K,假设实验中共有两个组分A和B,其中A组分为弱滞留性成分,B组分为强滞留性成分,所对应的亨利常数分别为KA和KB,将两个K值标注于正方形平面的对角线上,那么平行于纵坐标且通过KA的直线与平行于横坐标且通过KB的直线将构成一个三角形区域,该三角形区域即为理论上可以分离A和B两组分的操作区间。依据式(1)及管柱设计形态分别求得第2段区段和第3段区段的m值,即分别为m2与m3值,然后以m2为横坐标,m3为纵坐标将每组操作条件标在平面上。依据实验结果则可推算出实际可分离A和B两组分的操作区间,以下将以虚线三角形表示。

式中:t为SMB单柱实验中组分的保留时间;t0为接管柱后系统的死时间;td为不接管柱时系统的死时间;εt为管柱的孔隙度。
2 结果与分析
2.1 超临界萃取条件优化结果
如图2所示,A组仅在萃取的前0.5 h可以得到讯号较强的萃取物,但峰响应低,目标物含量低,因此单独使用超临界二氧化碳无法有效地萃取灵芝子实体中的三萜类化合物。对比图2中A、B组可发现,两者的HPLC图谱的信号数量与峰形极其相似,说明此两种萃取方式所得到的产物基本一致。B组中各组分的响应值明显高于A组,表明添加乙醇作为夹带剂更能有效地萃取出灵芝三萜类化合物。这可能是因为灵芝三萜类化合物的极性偏高,更易溶于添加乙醇溶液的超临界二氧化碳中。因此,后续SMB实验中的进料选择B组方式进行萃取。
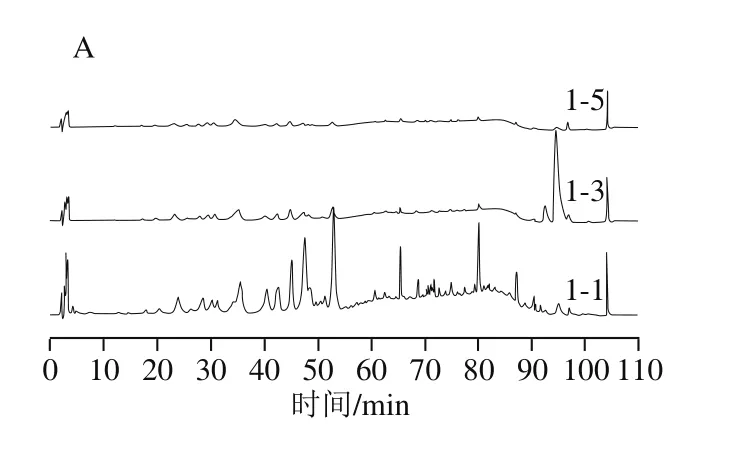
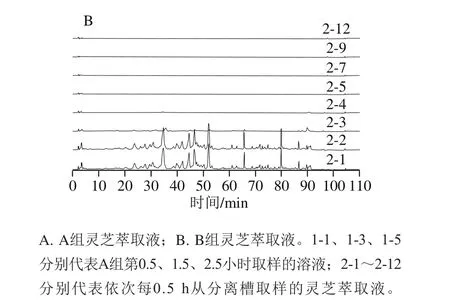
图2 A组(不添加夹带剂)和B组(添加乙醇为夹带剂)萃取液的液相色谱图Fig. 2 Liquid chromatograms of extracts A (without co-solvent) and B(with co-solvent)
2.2 粗萃物中灵芝三萜类化合物含量分析
由图2B可看出,萃取2 h后大部分物质已基本萃取完全。取萃取前3 h中每0.5 h所收集的萃取液进行定量浓缩,干燥后可得到每0.5 h粗萃物的量,再根据目标物的浓度可算出每0.5 h粗萃物中目标物的质量分数,如表2所示。计算结果表明2-4之后的样品中灵芝酸A、灵芝酸F及灵芝醇B的质量分数均为0或极少,即萃取2 h后目标组分已萃取完全,这与图2B结果一致。将前2 h收集到的萃取液混合,挥干溶剂共收集得到1.30 g,代表每千克子实体可以萃取到9.49 g的粗萃物。同时依据表2所得质量浓度,可计算得出3 种目标成分在粗萃物中的质量分数:灵芝酸A为4.50%,灵芝酸F为3.39%,灵芝醇B为0.29%。

表2 超临界二氧化碳混合乙醇萃取灵芝三萜类化合物含量Table 2 Contents of triterpenoids extracted by supercritical CO2 extraction using ethanol as co-solvent
2.3 SMB初始参数确定
单柱实验中,不同流动相配比(95%乙醇溶液与0.01%盐酸溶液体积比为95∶5、90∶10、85∶15、50∶50、45∶55、40∶60)的分离色谱图如图3所示。通过色谱图可见,乙醇溶液比例高于85%时,可有效地将样品中的所有物质脱附下来,但是当乙醇溶液比例低于50%时,便无法将全部样品脱附。因此若使用低比例乙醇溶液的流动相,在组态设计上需要加入润洗的操作,且润洗操作时的流动相需提高乙醇溶液的比例,才能使固定相成功再生。依据这些单柱层析图谱结果,本研究设计出2 组SMB分离实验,通过去除粗萃液中高极性和低极性杂质,达到纯化灵芝三萜类化合物的目的。其中,第1组SMB的实验以加入0.01%盐酸的乙醇溶液作为流动相,第2组SMB的分离则以体积比例为40∶60的乙醇溶液与盐酸(0.01%)混合溶液作为流动相。

图3 不同流动相的单柱分离色谱图Fig. 3 Chromatograms for single column separation with different mobile phases
2.4 第1组SMB分离实验与结果
传统的SMB共有4 个区段组成,其中第4区段主要是利用固体吸附剂将残留在移动相中的弱滞留性物质成分清除干净,避免被移动相带出床体之外继续循环而导致污染。由于本实验中灵芝粗萃液中的成分种类多而复杂,因此在组态设计上选择开放式系统,并且删除第4区段,以不回流移动相的方式解决弱滞留成分可能循环累积在系统的问题,如图4所示。设定SMB组态为2/3/3,其中共有2 个入口,即进料端(F端)、移动相端(D端),以及2 个出料口,即萃出液端(E端)、萃余液端(R端)。设定各进出口端的流速为D端5 mL/min,F端0.5 mL/min,E端2.083 mL/min,R端3.417 mL/min。

图4 第1组SMB实验的管柱组态设计Fig. 4 SMB column configuration design for experimental group 1
当系统达稳态操作后,在2个出口端收集样品并分析。在固定流速的情况下,实验进行了不同切换时间的测试,由表3可知,在切换时间为3.4 min时移除强滞留性的杂质效果最佳。且在该条件下,3 种目标物的质量分数相应提高,灵芝酸A提高至5.70%,灵芝酸F提高至4.17%,灵芝醇B提高至0.85%。由于所增加的质量分数有限,推测是因为低极性杂质的含量不多,所以指标成分质量分数增加有限。
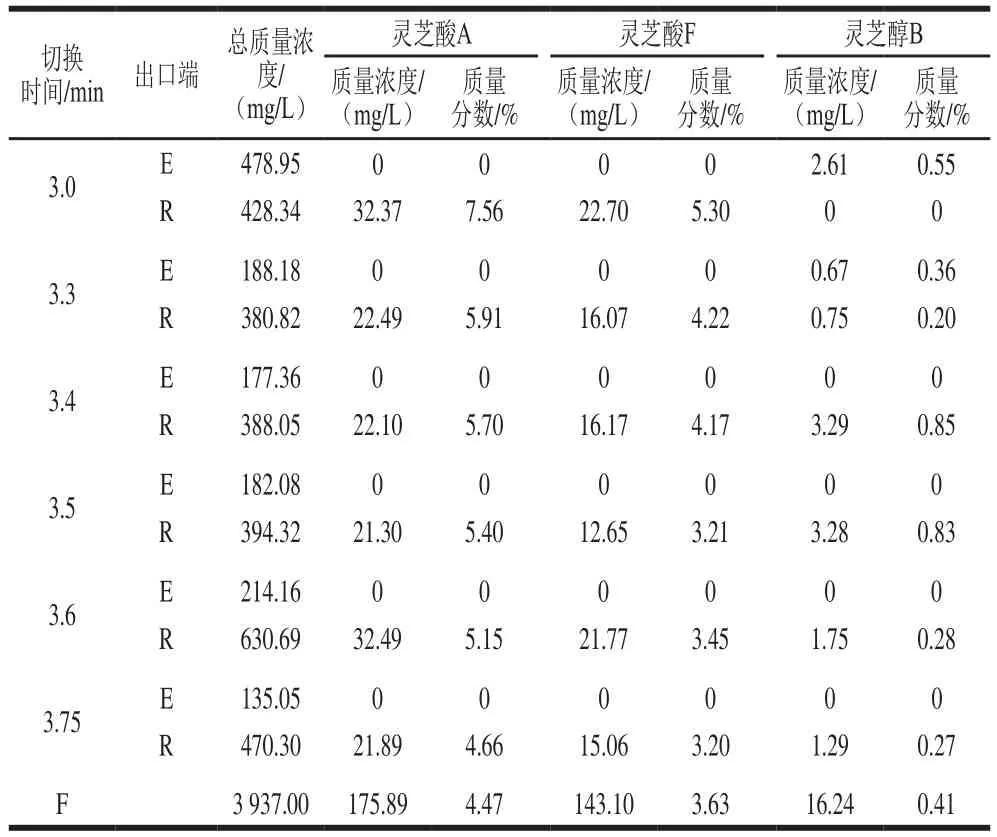
表3 不同切换时间的SMB分离结果(第1组)Table 3 Results of SMB separation at different switching times in experimental group 1
依据式(1)分别求得m2与m3值,然后以m2为横坐标,m3为纵坐标绘图所构成的平面图称其为(m2,m3)平面。如果将SMB实验所进行的操作条件分别标示在(m2,m3)平面上,如图5所示,图中实线构成的直角三角形代表三角形理论计算能够分离低极性杂质的操作区间,其中三萜类化合物与杂质的等温吸附常数分别为0.743与1.107。依据表3可见,在切换时间周期大约为3.3~3.5 min之间,可以有效移除弱滞留性成分,并依循Yu Hongwei等[33]提出建立最佳操作条件的方法,本研究绘制出图5虚线所构成的三角形。该虚线构成的三角形顶点坐标为(0.759,1.014),如果在此操作条件下进行,假设切换时间设为1.0 min,那么其各出入口端的流量设定分别为D端12.6 mL/min,F端1.677 mL/min,E端2.389 mL/min,R端11.888 mL/min。若按照最佳条件的流速设定SMB各进出口的流速,以同样的进料质量浓度3 937 mg/L进行实验,那么可预测此SMB设备平均每天每升填料的处理量,即本SMB系统在移除低极性杂质时固定相的效率为0.061 kg/(L·d)。
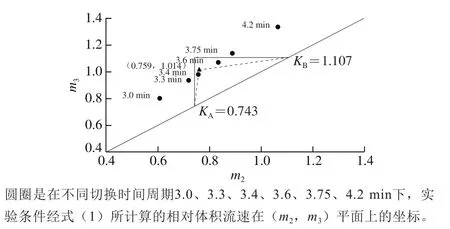
图5 第1组SMB分离实验三角形理论预测Fig. 5 Triangle theory prediction of SMB separation in experimental group 1
2.5 第2组SMB分离实验与结果
用1.3.1节B组方法所得到的萃取液进行浓缩,并取浓缩溶液400 mL,再加入600 mL的0.01%盐酸,过滤后得第2次进料溶液。如图6所示,本组SMB分离实验设定SMB组态为1-1-3/3,其中共有4 个入口,即进料端(F端)、移动相端(D端)、清洗端(Wash端)、润洗端入口(Rinse1端)以及3 个出料口,即萃余液端(R端),清洗端出口(W端),润洗端出口(Rinse 2端)。移动相使用乙醇-盐酸体积比40∶60,清洗端则使用添加0.01%盐酸的乙醇溶液,并设定各进出口端的流速为D端3.8 mL/min,F端0.3 mL/min,R端4.1 mL/min,Wash端5 mL/min,Rinse1端5 mL/min。
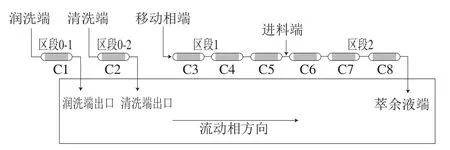
图6 第2组SMB实验的管柱组态设计Fig. 6 SMB column configuration design for experimental group 2
根据表4可知,在切换时间为34 min时,R出口端目标组分未有响应,但根据干质量结果可知R出口端分离出大量物质,但在本分析方法的检测波长下无法测出。同时根据Wash端的含量计算结果发现3 种目标物质量分数大幅度提高,其中灵芝酸A质量分数为19.34%,灵芝酸F质量分数为15.51%,灵芝醇B质量分数为0.74%。这代表大部分的三萜类化合物成分是高极性杂质,少部分为第1组SMB所分离出的低极性杂质。因此针对超临界流体萃取所得的粗萃物,提高三萜类化合物含量的重点在移除高极性的杂质。
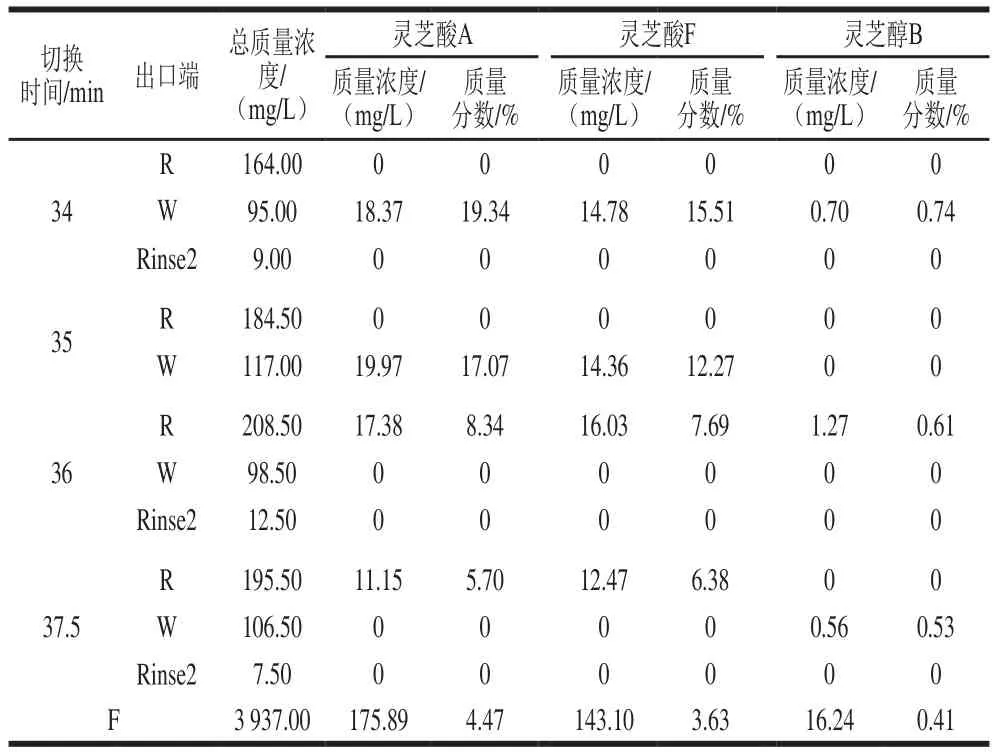
表4 不同切换时间的SMB分离结果(第2组)Table 4 Results of SMB separation at different switching times in experimental group 2
同样如果将第2组SMB分离实验所进行的操作条件分别标示在(m2,m3)平面上,如图7所示。根据表4结果可判定分离的切换时间周期在34~35 min之间,据此绘出图中虚线构成的三角形,并计算出其顶点坐标为(6.133,7.662)。如果在此操作条件下进行,假设切换时间设定为1.0 min,那么其各出入口端的流量设定分别为D端64.8 mL/min,F端11.709 mL/min,E端13.712 mL/min,R端62.796 mL/min,并以进料浓度3 937 mg/L进行实验,预测此本SMB系统在移除高极性杂质时固定相的效率为0.423 kg/(L·d)。

图7 第2组SMB分离实验三角形理论预测Fig. 7 Triangle theory prediction of SMB separation in experimental group 2
3 结 论
SFE在加入乙醇作为夹带剂后可有效从灵芝子实体萃取出灵芝三萜类化合物,且萃取物中三萜类化合物含量高。SMB的实验显示:使用酸性流动相可以有效移除非三萜成分;灵芝粗萃液中高极性杂质含量较低极性杂质含量多。SMB可有效地移除灵芝粗萃液中的杂质,灵芝三萜类化合物从进料溶液中质量分数为8.51%提高至35.59%,大幅提高了灵芝酸的质量分数。辅以三角形理论所建立的最佳操作条件,预测了本SMB系统在移除低极性杂质时固定相的效率为0.061 kg/(L·d),而移除高极性杂质时固定相的效率为0.423 kg/(L·d)。
