固相萃取-超高效液相色谱-串联质谱法测定饮用水中双酚A 和双酚S
2019-10-29杨金泉贺小敏施敏芳李永蓉朱友林
杨金泉,贺小敏,*,施敏芳,陈 浩,李永蓉,朱友林
(1.湖北省环境监测中心站,湖北 武汉 430072;2.华中农业大学理学院,湖北 武汉 430070;3.长江大学资源与环境学院,湖北 武汉 430100)
双酚A(bisphenol A,BPA)是世界上使用最广泛的工业化合物之一,主要用于生产聚碳酸酯、环氧树脂、聚砜树脂等多种高分子材料,在食品包装和容器内壁涂装等方面应用广泛,在食品的加工、运输、贮藏等过程中很容易迁移至食品中[1]。近年研究表明,BPA是一种外源性环境激素,具有拟雌激素活性,微量甚至痕量浓度的量也可能对动物生理状况、生殖系统以及胎儿发育造成不良影响[2],含有BPA的塑料婴儿奶瓶在美国、加拿大、日本等国家已禁止使用和销售[3]。目前,人们常用双酚S(bisphenol S,BPS)作为BPA的替代品[4],它是BPA分子中的丙烷基被砜基取代而得到的衍生物(图1),但有研究表明BPS具有与BPA类似的生物毒性,同样能对生物体产生雌性激素影响,也是一种内分泌干扰素[5]。GB 5749—2006《生活饮用水卫生标准》[6]规定饮用水BPA不得大于0.01 mg/L;GB 9685—2016《食品接触材料及制品用添加剂使用标准》[7]规定涂层中BPS的限定值,要求其特定迁移量不得超过0.05 mg/kg。但目前我国还没有饮用水中BPA、BPS标准检测方法,因此,建立灵敏高效的BPA和BPS分析方法具有重要意义。
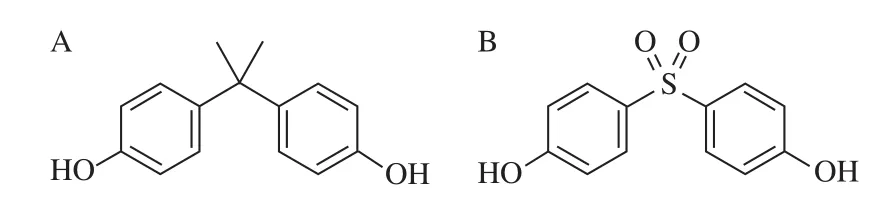
图1 BPA(A)和BPS(B)的结构式Fig. 1 Structures of BPA (A) and BPS (B)
由于饮用水中BPA和BPS含量通常较低,需要采用液液萃取[8-9]、固相萃取[10-12]和固相微萃取[13-14]等前处理方法进行富集,然后通过气相色谱[15]、液相色谱[16-17]、气相色谱-质谱联用[18-20]或液相色谱-质谱联用[21-24]等仪器进行测定。液相色谱定性不够准确、易产生假阳性,且灵敏度较低;气相色谱和气相色谱-质谱联用测定BPA和BPS往往需要对样品进行衍生化,操作繁琐、费时费力;液相色谱-质谱联用法定性准确、灵敏度高、无需繁琐的衍生化,在双酚类环境激素的分析中应用越来越多。目前,利用液相色谱-质谱联用法测定BPA的研究较多,但对BPS的检测报道较少。本实验建立了固相萃取-超高效液相色谱-串联质谱法同时检测饮用水中BPA和BPS的定量分析方法,并对湖北省内6 处饮用水源地水样展开应用分析,为饮用水中BPA和BPS的科学管控提供可靠的分析方法。
1 材料与方法
1.1 材料与试剂
BPA、BPS(纯度>98%) 德国Dr. Ehrenstorfer GmbH公司;甲醇、乙腈(均为色谱纯) 美国Tedia公司;氟化铵(色谱纯) 美国Fluka公司;Oasis HLB固相萃取柱(500 mg/6 mL) 美国Waters公司;0.22 μm微孔滤膜;水为Milli-Q超纯水;高纯氮气99.999%。
1.2 仪器与设备
1290超高效液相色谱仪、6460三重四极杆质谱仪(配置电喷雾电离源、ZORBAX Extend-C18反相色谱柱(100 mm×2.1 mm,3.5 μm)) 美国Agilent公司;VisiprepTMDL 12孔固相萃取装置(配有真空泵和虹吸管) 美国Supelco公司;TurboVap LV氮吹仪美国Biotage公司。
1.3 方法
1.3.1 标准溶液的配制
标准储备溶液的配制(100 mg/L):分别准确称取0.010 0 g BPA和BPS标准品,用甲醇定容至100 mL,4 ℃避光保存。
工作曲线的配制:准确移取BPA和BPS标准储备溶液各1 mL于100 mL容量瓶中,用甲醇配制成混合标准中间液,再以甲醇-水溶液(1∶1,V∶V)逐级稀释为不同质量浓度梯度(1.0、2.0、5.0、10.0、20.0、50.0、100.0、200.0 μg/L)的混合标准工作溶液,4 ℃避光保存。
1.3.2 样品采集和保存
在湖北省武汉(3 个水厂)、黄石(1 个水厂)、孝感(1 个水厂)、咸宁(1 个水库)地市6 个饮用水源地共设置12 个监测点位,采集入水口和出水口样品。样品采集和保存参考标准GB/T 5750.2—2006《生活饮用水标准检验方法 水样的采集和保存》,使用预先洗净烘干的棕色玻璃瓶保存样品,避光于4 ℃冷藏,在7 d内萃取,萃取后的样品应避光于4 ℃冷藏,40 d内分析完毕。
1.3.3 样品前处理
依次用10 mL甲醇和10 mL超纯水对HLB固相萃取柱进行活化,备用。量取500 mL水样以3~5 mL/min的流速通过活化后的HLB小柱,然后用10 mL 10%甲醇溶液淋洗固相萃取小柱后继续抽吸1 h。用10 mL纯甲醇以1 mL/min的流速洗脱,洗脱液于37 ℃恒温水浴并用氮气浓缩至0.5 mL,然后以超纯水定容至1 mL,经0.22 μm微孔滤膜过滤后进行超高效液相色谱-串联质谱分析。
在分析样品的同时做全程序空白实验,即按照上述实验过程以超纯水代替水样完成全部分析流程,检查分析过程中是否存在污染。
1.3.4 仪器工作条件
1.3.4.1 色谱条件
流动相为1 mmol/L氟化铵溶液(A)和乙腈(B);流速0.3 mL/min;进样量10 μL;柱温30 ℃。流动相梯度洗脱程序见表1,后运行3 min。

表1 梯度洗脱程序Table 1 Gradient elution program of ultra performance liquid chromatography
1.3.4.2 质谱条件
电喷雾电离负离子模式,监测模式为多反应监测(multiple reaction monitoring,MRM)模式,离子源温度350 ℃,干燥气流速10 L/min,鞘气流速11 L/min,雾化器压力45 psi,毛细管电压3 500 V。质谱MRM模式参数见表2。
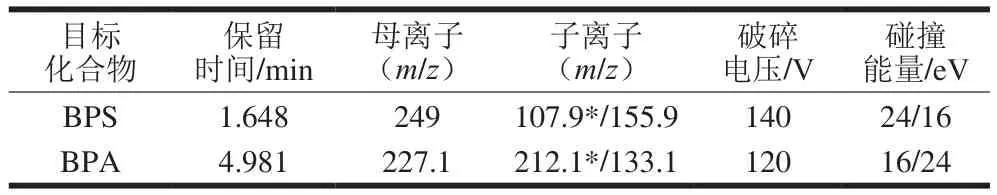
表2 BPA和BPS的质谱采集参数Table 2 Mass spectrometric parameters for BPA and BPS
1.3.5 方法学验证
用甲醇-水溶液(1∶1,V/V)配制混合标准溶液系列,以化合物质量浓度为横坐标,色谱峰面积为纵坐标作标准曲线。方法检出限依据标准HJ 168—2010《环境监测分析方法标准制修订技术导则》,按照样品分析的全部步骤,对低质量浓度加标样品进行不少于7 次平行测定,计算标准偏差和方法检出限。
分别取500 mL空白样品,添加低、中、高3 个水平的BPA和BPS混合标准溶液,进行添加回收实验,计算相对标准偏差(relative standard deviation,RSD)和加标回收率。
1.4 数据处理
采用Origin 8.0软件对实验数据进行处理,计算最大值、最小值、平均值和RSD,并根据化合物的响应值或回收率数据作出相应色谱图、柱形图等图表。
2 结果与分析
2.1 分析条件的优化
2.1.1 质谱参数的优化
分别将1 mg/L BPA和BPS标准溶液注入质谱仪进行质谱参数的优化。先通过全扫描确定化合物的母离子和正负离子采集模式,再进行选择离子监测优化其碎裂电压及毛细管电压使母离子的响应最大,然后对母离子做子离子全扫描,获得碎片离子信息,选择丰度较高的2 个特征碎片离子作为定量离子和定性离子,优化碰撞能量使其响应最大,最后得到该化合物的2 个MRM离子对及相应的质谱参数(表2)。在该条件下获得的BPA和BPS标准溶液的MRM色谱图如图2所示。BPA和BPS均在负离子模式采集时响应较高。这主要是由于BPA和BPS均含有酚羟基,易失去一个H+产生[M-H]-母离子,即负离子模式。
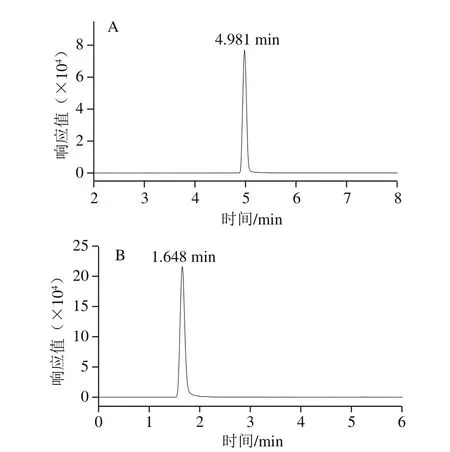
图2 BPA(A)和BPS(B)标准溶液的多反应监测模式色谱图Fig. 2 Multi-reaction monitoring chromatograms for BPA (A) and BPS (B) in a standard solution
2.1.2 流动相的优化
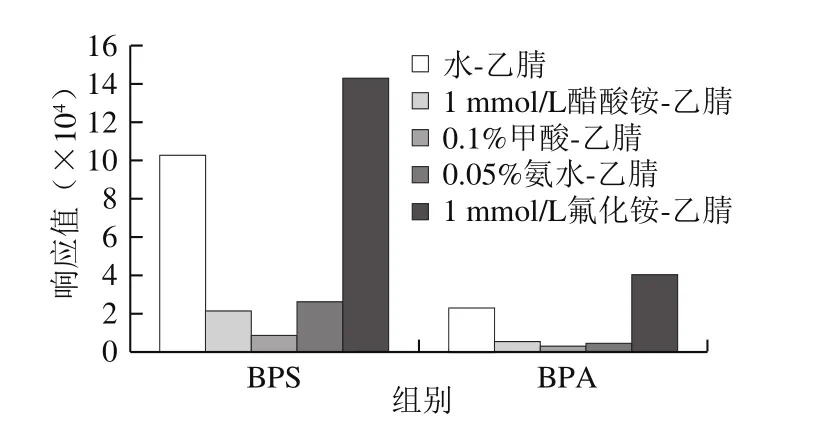
图3 不同流动相下BPA和BPS的响应值比较Fig. 3 Comparison of responses for BPA and BPS in different mobile phases
考察BPA和BPS在酸性、中性、碱性及不同缓冲盐流动相条件下的分离效果和响应值大小。因甲酸、氨水、醋酸铵、氟化铵等化合物挥发性较强,不易在色谱柱和质谱中残留,实验主要采用以上几种化合物调节流动相pH值和离子强度,对比不同浓度的氨水-乙腈、水-乙腈、甲酸-乙腈、醋酸铵-乙腈、氟化铵-乙腈等作为流动相时,BPA和BPS的响应值大小,如图3所示。结果表明:这2 种双酚类物质均在氟化铵-乙腈条件下响应值最高,峰形最好。进一步考察不同浓度(0.5、1 mmol/L和2 mmol/L)氟化铵-乙腈作为流动相对BPA和BPS响应值的影响。结果显示,随着氟化铵浓度增大,两种双酚类物质响应值均有所增加,2 mmol/L氟化铵与1 mmol/L氟化铵时,其响应情况相当,考虑到氟化铵有一定的腐蚀性,浓度过大可能对色谱柱有一定的损害,故最终确定1 mmol/L氟化铵溶液-乙腈为流动相。
2.2 前处理条件的优化
2.2.1 固相萃取柱的选择
由于BPA和BPS属弱极性化合物,通常采用C18柱、HLB柱等反相柱对其进行富集,实验比较了相同规格的C18、HLB、NH2固相萃取柱对BPA和BPS的回收率情况(图4)。结果表明,NH2柱对BPA和BPS的富集效果很差,尤其是对BPA基本无保留;C18柱对两种物质的保留能力一般,回收率低于62%;而HLB柱对各目标物的保留能力相对较好,回收率分别为85%和97%。这可能是由于BPA和BPS极性偏强,但电离程度弱,主要呈分子形态,不适宜采用NH2柱这类离子交换型填料,而C18柱更适合富集非极性化合物,萃取原理为疏水效应。相比较而言,HLB柱中的填料是一种亲水亲脂型聚合物,可富集的化合物极性范围较宽,因此HLB柱对BPA和BPS均有较好的萃取效果。
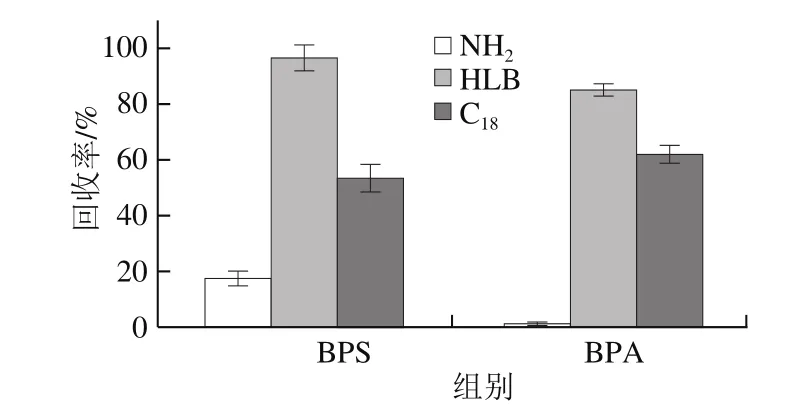
图4 不同固相萃取柱对BPA和BPS回收率的比较Fig. 4 Comparison of recoveries for BPA and BPS on different SPE columns
2.2.2 淋洗液的选择
为去除基质干扰,可在洗脱前对固相萃取小柱进行淋洗,通常在淋洗液中添加一定比例的甲醇,去除部分强极性或水溶性杂质,但同时也可能造成目标物的损失[25]。本实验考察不同体积分数(5%、10%、15%、20%)的甲醇溶液作为淋洗液时,样品基质去除情况及回收率情况。结果表明,当甲醇体积分数为10%时,BPA和BPS回收率分别为84%和95%,而当甲醇体积分数增至15%时,BPA回收率下降较多,仅为67%。因此要保证回收率的同时,最大程度去除基质干扰,应选择10%的甲醇溶液为淋洗液。
2.2.3 洗脱液和体积的选择
根据文献报道,使用C18和HLB小柱萃取BPA和BPS时,大多采用甲醇和乙腈作为洗脱剂[23,26-29]。因此实验比较了甲醇和乙腈对两种目标物的洗脱情况,结果发现两者洗脱效果相当,考虑到乙腈沸点高(甲醇沸点64.7 ℃、乙腈沸点81.6 ℃),浓缩时间长,因此选择甲醇为洗脱剂。考察不同体积的甲醇对两种化合物的洗脱情况,作出流出曲线(图5)。由图5可见,BPA和BPS在洗脱体积为10 mL时回收率已趋于稳定,因此方法最终选择洗脱剂用量为10 mL。
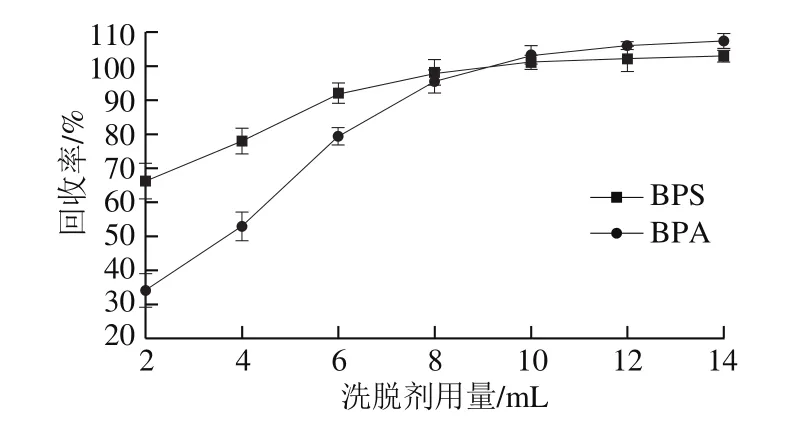
图5 BPA和BPS的流出曲线Fig. 5 Elution curves of BPA and BPS
2.3 线性关系、精密度、准确度和检出限
分别以BPA和BPS峰面积为y轴,化合物质量浓度为x轴绘制标准曲线,得到线性回归方程,结果见表3。BPA和BPS在1~100 μg/L范围内线性关系良好,相关系数均超过0.995。将两种双酚类化合物标准样品添加至超纯水中(BPS 2 ng/L、BPA 10 ng/L),按照上述条件进行前处理,平行测定7 份样品,按照3.143 倍标准偏差获得两种化合物的方法检出限,与已有报道相比,本方法检出限低1~2 个数量级,能够满足饮用水中痕量BPA和BPS的分析测定要求。
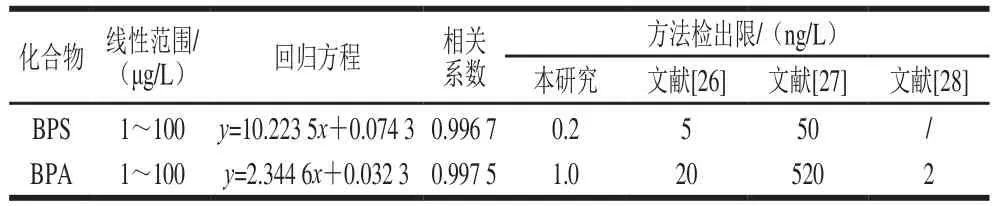
表3 BPA和BPS的标准曲线和方法检出限Table 3 Linear equations and LODs for BPA and BPS
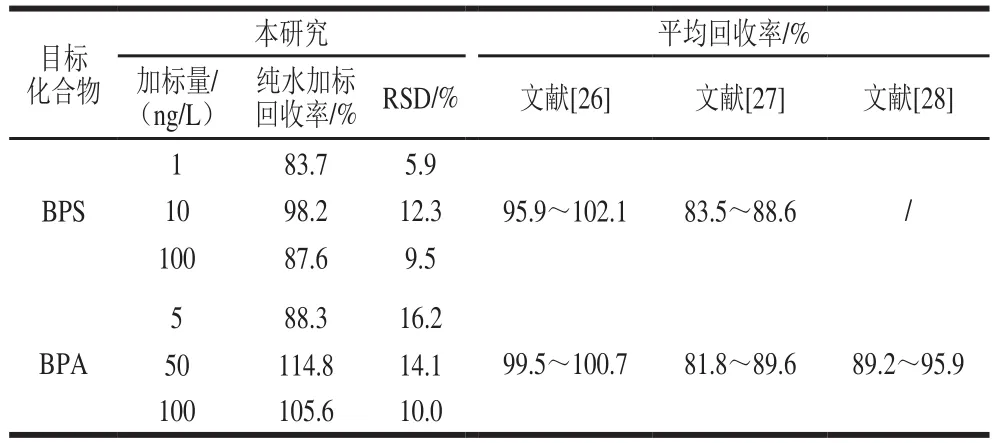
表4 BPA和BPS的加标回收率和RSD(n= 3)Table 4 Recoveries and RSDs of BPA and BPS (n= 3)
按优化的方法进行目标化合物的加标回收率实验,在纯水中加入低、中、高3 个水平的BPA和BPS标准样品,每个添加水平设置3 个平行样,计算加标回收率和RSD(表4)。结果表明,BPS和BPA加标回收率分别为83.7%~98.2%和88.3%~114.8%,RSD为5.9%~12.3%和10.0%~16.2%。此外,实验还对实际水样进行了基体加标实验(表5),两种化合物的加标回收率分别为80.9%~96.2%和74.8%~118.0%。比较发现本研究纯水和实际水样中BPA和BPS的加标回收率范围与已有文献报道相当,表明本方法准确度较好,适合于饮用水的分析测定。

表5 BPA和BPS的实际样品分析结果Table 5 Contents of BPA and BPS in drinking water samples
2.4 实际样品的分析
采用建立的固相萃取-超高效液相色谱-串联质谱检测方法,对湖北省地市内6 个饮用水源地共12 个点位的水样中BPA和BPS进行分析,其中5 个点位有不同程度的BPA检出(表5),质量浓度范围为2.3~14.8 ng/L,BPS均未检出,空白样品和实际样品色谱图见图6。与已有文献[29-31]分析比较发现,本研究饮用水样中BPA质量浓度范围与已有文献报道基本一致,而饮用水中BPS鲜见有报道检出。
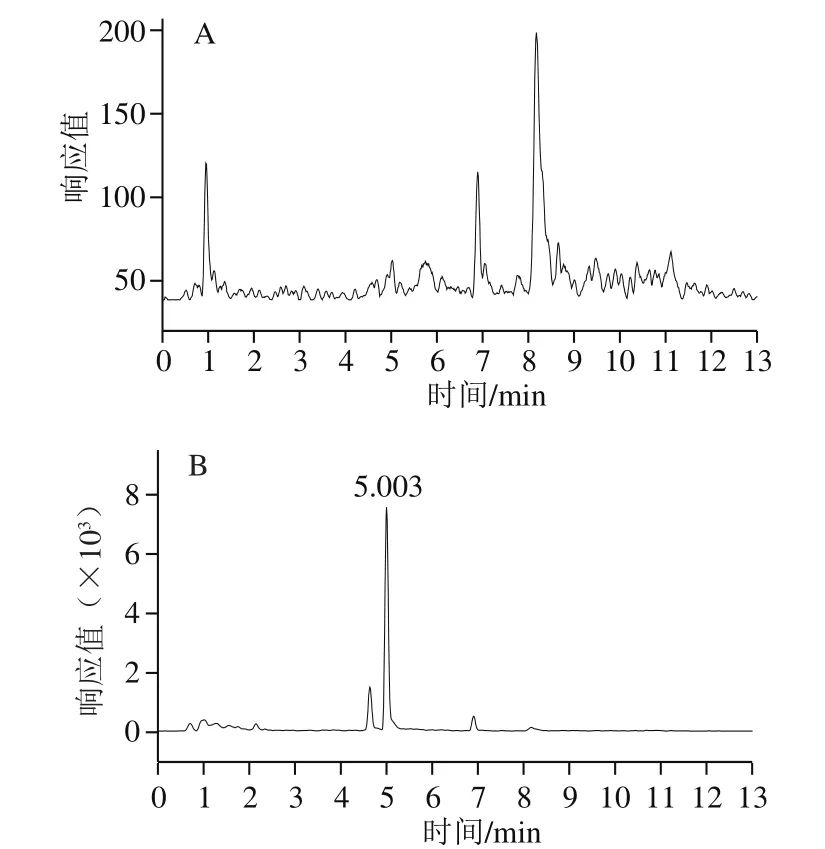
图6 BPA空白样品(A)和实际水样(B)MRM图Fig. 6 Multi-reaction monitoring chromatograms for BPA in a blank sample (A) and water sample (B)
3 结 论
本研究采用HLB固相萃取样品中BPA和BPS,经超高效液相色谱-串联质谱法定量,建立了饮用水中痕量BPA和BPS的检测方法。利用该方法对湖北省内6 处饮用水源地样品进行BPA和BPS含量测定,结果表明,本方法操作简单高效、定性准确、精密度好、检出限低,远低于GB 5749—2006规定的BPA限定值0.01 mg/L,满足饮用水检测限量要求。
