LDLR基因突变致家族性高胆固醇血症家系分析
2019-10-27刘军刘芳
刘 军 刘 芳
家族性高胆固醇血症 (familial hypercholesterol-emia,FH)是一种以血浆总胆固醇(total cholesterol,TC) 和低密度脂蛋白胆固醇(low density lipoprotein cholesterol,LDL-C) 水平增高,机体不同部位皮肤/肌腱散发大小不等的黄色瘤、早发动脉粥样硬化、早发冠心病为特征的常染色体遗传性疾病, FH有杂合子型和纯合子型两种,发生率约1/500~1/200和1/30万~1/16万[1~3]。杂合子型主要是低密度脂蛋白受体(low density lip-oprotein receptor,LDL-R)、载脂蛋白 B(apolipoproteinB,ApoB)、枯草溶菌素转化酶 9(proprotein convertase subtilisin/kexin type 9,PCSK9)的功能改变,纯合子型主要是 LDL-R 衔接因子蛋白 1(low density lipopro-tein receptor adaptor protein 1,LDLRAP1)功能改变所致[4]。
本研究报道了一个高胆固醇血症家系4例患者的临床表现、生化检测及基因测定结果,证实该家系患者均存在LDL-R基因突变,现报道如下。
对象与方法
1.研究对象:先证者:4岁,女性,因发现身体不同部位皮肤出现大小不等的黄色凸起物就诊。患儿系G3P2,孕40周顺产娩出,羊水、胎盘、脐带无异常,阿氏评分1、5、10min均为10分,无窒息抢救史。出生后母乳喂养,母自诉患儿平素身体健康,饮食无特殊嗜好。大约半年前患儿四肢皮肤多处出现深浅及大小不一的黄色凸起物,压之不退色,不伴发热,无心前区不适。无母孕期射线和服药物史,患儿有一个姐姐11岁,一个弟弟1岁,二人体健,无皮肤黄色凸起物。父母非近亲婚配,否认有遗传病家族史。先证者入院查体:生命体征平稳,神志清,体重18kg,身高1.1m, 呼吸平稳,双肺听诊无异常,心音有力,无杂音,腹部脊柱无异常,双腿膝关节屈侧肌腱处、小腿踝关节伸侧屈侧肌腱处、双肘关节屈侧肌腱处可见大小不一的黄色瘤(图1), 双眼有明显角膜弓,心电图、超声心动图正常。血TC 16.40mmol/L,高于正常3倍余,LDL-C 15.06mmol/L,高于正常近5倍,载脂蛋白B 2.78g/L高于正常2倍余(表1)。其他成员: 先证者父母、弟弟皮肤或肌腱未发现黄色瘤,也无其他临床症状。心电图、心脏彩超正常,但3个人血TC、LDL-C、载脂蛋白B均有不同程度的增高,只是增高幅度低于先证者。先证者姐姐临床无症状,上述指标也无增高。
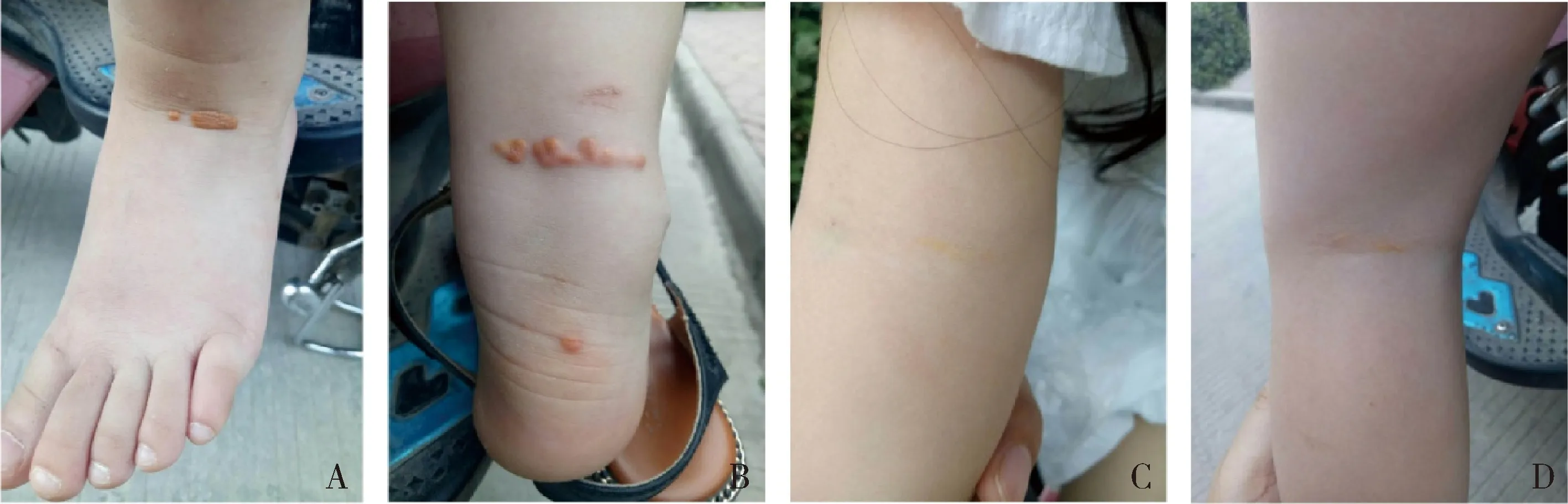
图1 先证者踝关节伸侧屈侧肌腱附近、肘关节肌腱处、膝关节肌腱处大小不一的黄色瘤A.伸侧肌腱附近;B.屈侧肌腱附近;C.肘关节肌腱处;D.腱关节肌腿处
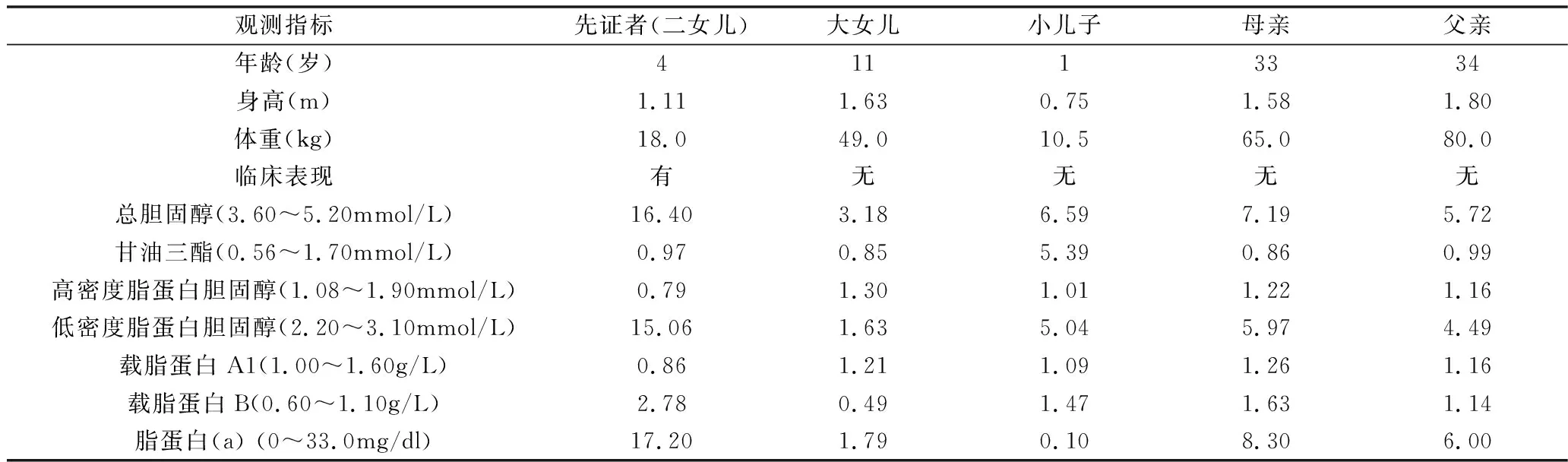
表1 先证者一家5口年龄、身高、体重及血脂检测水平
2.外周血基因组DNA的提取:采集先证者、其父亲、母亲、姐姐、弟弟外周血各2ml EDTA抗凝,用QIAamp全血DNA提取试剂盒(德国Qiagen 公司)按其指南提取基因组DNA。以上获得笔者医院医学伦理学委员会讨论通过后,家长签署了知情同意书。
3.医学外显子测序:DNA送第三方检验公司-北京迈基诺基因公司,对先证者及父母行医学外显子测序。使用Hiseq2000标准流程测序(二代测序仪:Illumina NextSeq500, 美国Illumina公司)。
4.序列比对,变异注释和危害性预测:通过Q20标准对原始数据进行数据过滤得到Clean data,使用BWA比对软件,将所有的Clean reads 比对到人参考基因组(HG19);将分析得到的SNP和INDEL通过ANNOVAR软件进行注释。筛选掉正常人数据库中频率<0.05的突变位点, 用MutationTaster和GERP++软件进行致病性预测和保守性预测,剪切位点的改变用SPIDEX软件分析其致病性。
5.一代测序(Sanger法)验证:筛选到的候选变异位点用PCR和Sanger测序验证(ABI3730xl测序仪,美国Applied Biosystems 公司)。最后对先证者姐姐、弟弟行致病突变点的验证。
结 果
1.先证者家系医学外显子测序分析:先证者样本分析到 LDL-R 基因有 2个杂合突变:(1)在LDL-R基因的第5个外显子上有一个杂合突变,即c.727T>A(编码区第 727 号核苷酸由胸腺嘧啶变异为腺嘌呤),导致氨基酸改变 p.C243S(第 243 号氨基酸由半胱氨酸变异为丝氨酸),为错义突变。该变异在正常人群数据库无携带频率。蛋白功能预测软件 SIFT、PolyPhen_2、REVEL的预测结果均为有害。在 HGMD专业版数据库中未见报道。经家系验证分析,先证者之父该位点杂合变异,先证者之母该位点无变异。对先证者姐姐和弟弟进行验证,结果显示先证者弟弟该位点同先证者父亲为杂合变异,先证者姐姐该位点无变异(图2)。(2)在LDL-R基因的第7个外显子上有一个杂合突变,即c.1003G>T(编码区第 1003 号核苷酸由鸟嘌呤变异为胸腺嘧啶),导致氨基酸改变 p.G335C(第 335号氨基酸由甘氨酸变异为半胱氨酸),为错义突变。该变异在正常人群数据库无携带频率。蛋白功能预测软件 SIFT、PolyPhen_2、REVEL的预测结果分别为有害、有害、有害。在 HGMD专业版数据库中未见报道。经家系验证分析,先证者之父该位点无变异,先证者之母该位点杂合变异。对先证者姐姐和弟弟进行验证,结果显示先证者姐弟俩该位点均无变异(图2)。
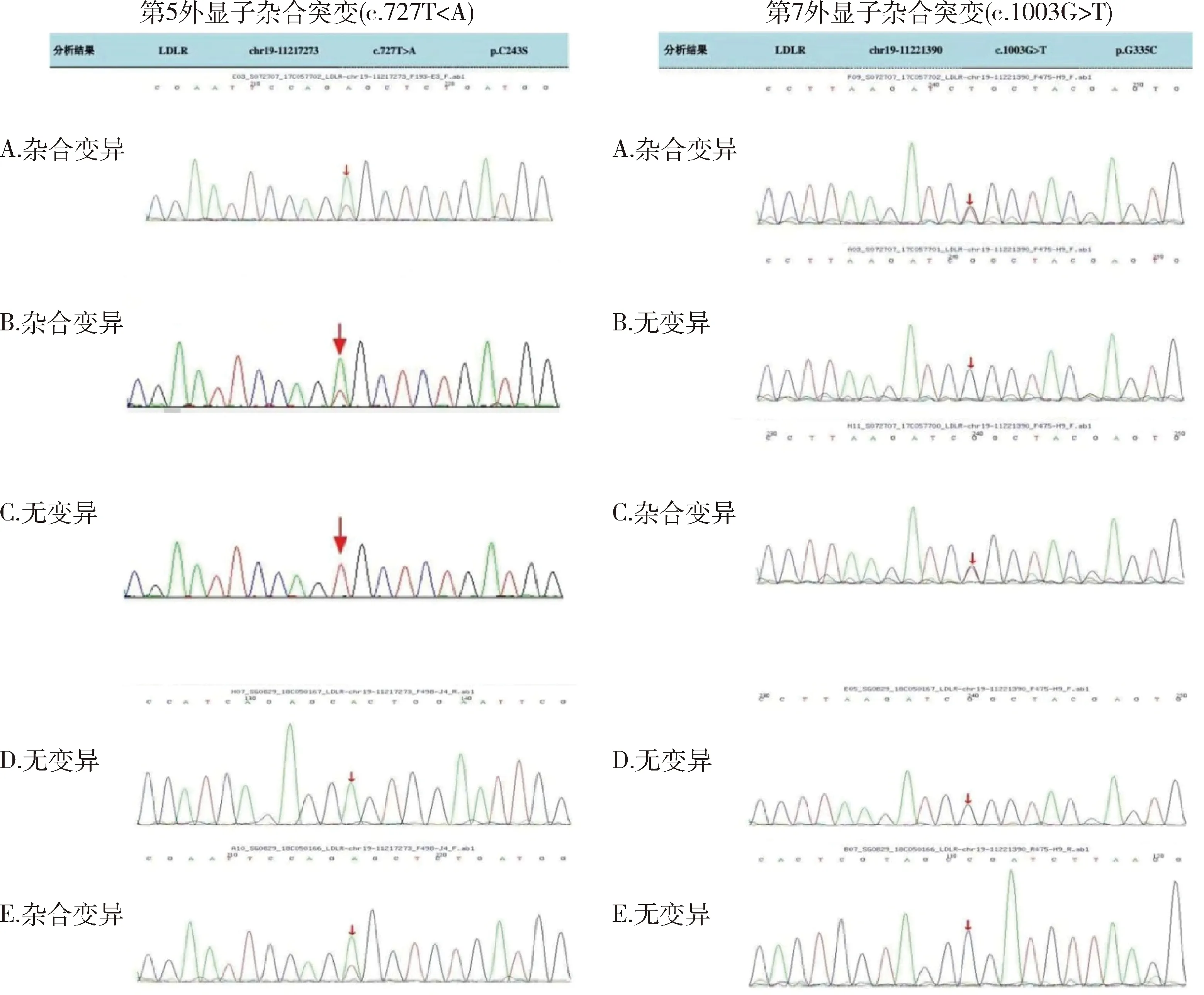
图2 先证者、父母、姐姐及弟弟DNA测序图A.先证者测序图;B.先证者父亲测序图;C.先证者母亲测序图;D.先证者姐姐测序图;E.先证者弟弟测序图;箭头指示为突变位点
2.随访:先证者父母经过4个月饮食控制加锻炼,TC、LDL-C有所下降,但仍然高于正常,后加用阿托伐他汀钙 20mg, 每晚1次,口服,2周后复查两人TC分别降至 4.2mmol/L和4.8mmol/L,LDL-C分别降至2.7mmol/L 和3.0mmol/L。先证者经过4个月饮食控制加锻炼,复查血TC 14.10mmol/L,LDL-C 11.74mmol/L,较前有所降低。由于患儿皮肤黄色瘤有所增多,家长十分焦虑。与家长协商后,试给患儿加用阿托伐他汀钙 5mg, 每晚1次,口服,2周后TC降至11.46mmol/L,LDL-C降至9.82mmol/L,继续维持现在剂量口服2周,必要时考虑增加到10mg, 每晚1次,口服,目前笔者还在密切随访先证者。先证者弟弟1岁余,主要吃奶和添加辅食,嘱少食内脏食物,最近1次复查TC为6.42mmol/L,LDL-C为4.06mmol/L。
讨 论
FH是一种常染色体显性遗传病,主要表现为血浆LDL-C水平异常升高,肌腱黄色瘤,以及早发冠心病。中华医学会心血管病学分会动脉粥样硬化及冠心病学组制定了中国FH筛查与诊治中国专家共识[5]。共识指出儿童FH诊断标准:未经治疗的血清LDL-C≥3.6mmol/L且一级亲属中有FH或早发冠心病。本先证者及弟弟血清LDL-C分别是15.06mmol/L和5.04mmol/L,结合家族史,符合儿童FH诊断标准。共识对成人FH诊断标准是:符合下列3条中的2条:①未经治疗的血清LDL-C≥4.7mmol/L;②皮肤/腱黄色瘤/脂性角膜弓(<45岁);③一级亲属中有FH或早发动脉粥样硬化性心血管疾病(ASCVD)患者。先证者父母血清LDL-C分别为4.49mmol/L和5.97mmol/L,结合家族史符合成人FH诊断标准。FH 可分为纯合子和杂合子两种类型,杂合子症状通常较轻,而纯合子症状严重,表现为多部位黄色瘤、发展迅速的全身动脉粥样硬化,青少年时期发生心肌梗死甚至死亡。本家系中先证者LDL-R基因存在2个复合杂合变异,属于纯合子,变异的基因分别来自父母,其他家庭成员除了姐姐正常,LDL-R基因仅存在1个位点的突变,属于杂合子。
从临床上看,先证者血清LDL-C水平远高于正常范围,也高于其他家庭成员,这点也符合纯合子的特点,胆固醇代谢涉及了许多蛋白质和途径,LDL分解代谢是该过程中的关键点,参与其中的蛋白质功能发生任何缺陷都可以产生FH。其中起主要决定因素的为LDL-R,占FH病例的80%~85%,载脂蛋白B占5%~10%的病例,枯草溶菌素转化酶9占 2%,LDL-R衔接因子蛋白占不到1%的病例,其余的少见[4]。目前文献报道发现 LDL-R、载脂蛋白B、枯草溶菌素转化酶9、LDL-R衔接因子蛋白、固醇调节元件结合蛋白2、三磷酸腺苷结合盒转运体、胆固醇 7α-羟化酶这7 种基因突变可导致 FH,我国目前仅发现了前3种基因突变[3]。
LDL-R基因位于染色体19p13.2,由 18个外显子和 17个内含子组成, 包含 5个不同的功能域,当外显子发生突变可使 LDL-R相应的功能域不同程度受损, 导致LDL-R的功能障碍。文献报道LDL-R基因突变位点遍布启动子, 内含子及18个外显子中, 目前世界范围已从 FH患者中检测出了3000多种突变, 包括缺失、插入、无义突变、错义突变、拷贝数变异等[4,6~8]。
LDL-R基因突变为 FH 主要病理基础,LDL-R是一种富含半胱氨酸的839个氨基酸的蛋白质,是一种细胞表面糖蛋白,通过受体介导的内吞作用特异性结合并内吞含载脂蛋白B 100或载脂蛋白 E的脂蛋白,在胆固醇体内平衡中起重要作用。LDL-R基因突变导致细胞膜表面 LDL-R结构功能异常, 不能有效代谢体内LDL, 导致LDL因清除障碍而在血液或其他组织中过度淤积引发高胆固醇血症[6,9,10]。
据相关文献报道,中国FH 患者 LDLR基因突变均发生在 1~17 外显子,第 18 外显子突变尚未发现[11]。Chiou等[12]统计发现,在我国汉族人群中目前存在143种不同LDL-R突变类型(134种位点突变及9种基因重组)。2015年Jiang等[13]对中国 FH 人群基因情况予以概述,发现 LDLR 突变多位于外显子 4、9、13 及 14,主要是错义突变,占60.3%,C308Y、H562Y 和 A606T 突变最为常见。其中,我国内陆北方地区 A606T 突变最常见,内陆南方地区 W462X 突变最常见,台湾地区 C308Y 最常见。2017 年 Li 等[14,15]对全国范围内 245 例 FH 患者进行基因突变分析发现,在单基因突变FH患者中,LDL-R基因突变占68.2%,多位于外显子12、4、9。
本研究中FH先证者LDL-R基因有2个杂合突变(复合杂合子),一个位于第5号外显子,一个位于第7号外显子,分别来自父母,属于纯合子。这两个突变c.727T>A (p.C243S)和c.1003G>T (p.G335C)均为错义突变,在正常人群数据库无携带频率,蛋白功能预测软件 SIFT、PolyPhen_2、REVEL的预测结果均为有害,上述变异在 HGMD专业版数据库中未见报道,属于新发突变。由于先证者本身存在黄色瘤、高胆固醇和高低密度脂蛋白胆固醇,分析该基因突变为致病性突变。先证者父母、弟弟各存在1个基因突变,属于杂合子,3个人同样存在血高胆固醇和高低密度脂蛋白胆固醇,只是程度较纯合子的先证者轻。先证者姐姐临床生化检验正常,基因测序未见LDL-R基因突变,符合共分离特征,因此可以判断本家系中存在的FH致病因素是LDL-R基因突变所致。
为提高 FH 诊断率,应尽早对 FH 予以筛查,方法有人群筛查、选择性筛查、家族级联筛查、脂质级联筛查、基因级联筛查、反向级联筛查等。Starr 等[16]建议在年龄>2 岁的儿童中筛查 FH,再进一步对确诊儿童父母进行筛查,即进行反向级联筛查,这种筛查的优点是大多数国家儿童有常规体检,易实施。2017 年 Wu 等[17]在我国47例FH患者的1、2级亲属中进行反向级联筛查发现,总筛查检出比例为2.8例新诊患者/1 例先证者,证实我国反向级联筛查是高效的。
本研究报道的家系成员中,先证者作为儿童首先因为黄色瘤就诊,随后发现异常血脂增高,对其家中成员进行血脂筛查,结果父母、弟弟均不正常,仅姐姐正常。最后进行基因测定,证实家族中除了姐姐,其余成员均存在LDLR基因突变,且先证者为复合杂合突变,即存在两个位点的突变,属于纯合子,此检测结论也验证了先证者临床症状突出、血脂异常明显的特点。
FH 的治疗包括生活方式和饮食控制、药物治疗、脂蛋白血浆置换以及肝脏移植等[18]。成人 FH 患者一旦确诊应立即改变生活方式并开始药物治疗如他汀类药物、胆汁酸螯合剂等,对于纯合子患者及患有冠心病并对其他治疗无效的杂合子患者采取血浆清除。本研究先证者父母属于杂合子,临床没有症状,心电图和心脏超声均正常,治疗最初依靠饮食和锻炼,但血胆固醇不能降至满意,加用阿托伐他汀钙口服效果满意。本研究先证者为4岁多儿童,依靠调整饮食效果不理想,且皮肤黄色瘤增多,试加用阿托伐他汀钙5mg,每晚口服1次,目前血TC、LDL-C仍然比较高,由于患儿年龄小,笔者尚在密切随访治疗效果,必要时调整药物剂量为10mg,每晚口服1次。对于儿童患者,基于生长发育的考虑,无论纯合子型还是杂合子型 FH,2 岁前均不宜应用低脂饮食,因此先证者弟弟仅嘱少食动物内脏,其余未做特殊处理[4,19]。该研究不足之处是未对先证者皮肤黄色瘤予以病理学检查,原因是患儿父母坚决反对。
对一家族性高胆固醇血症家系进行医学外显子测定,结果显示该家系5人中的4人存在LDLR基因突变,分别是5号外显子c.727T>A 和(或)7号外显子c.1003G>T,这两种突变在正常人群数据库无携带频率,在 HGMD专业版数据库中未见报道,提示对临床出现不明原因皮肤黄色瘤,血胆固醇增高的儿童要积极寻找致病原因,对其亲属也应该积极进行相关检测,及早做出诊断,给予干预,避免疾病进展。
