应用富含GC区SNP分型的序列特异性PCR新方法检测子宫内膜癌特定位点C729T多态性
2019-10-24王彩凤
王彩凤,陈 葳,李 旭
(西安交通大学第一附属医院:1. 急诊科;2. 检验科;3. 转化医学中心,陕西西安 710061)
单核苷酸多态性(single nucleotide polymorphisms, SNPs)是个体中最常见的遗传变异,被广泛应用于群体遗传学、疾病相关基因定位与诊断、药物的开发与应用等研究中[1]。目前,SNP检测技术的传统方法包括限制性酶切片段长度多态性(restriction fragment length polymorphism, RFLP)[2-4]、单链构象多态性(single-strand conformational polymorphism, SSCP)[5]、等位基因特异性扩增(allele specific amplification, ASA)[6]等,有些检测过程较为繁琐,有些特异性较差等[5]。DNA芯片技术、变性高效液相色谱(denaturing high performance liquid chromatography, HPLC)、变性梯度凝胶电泳(denaturing gradient gel electrophoresis, DGGE)[6]、质谱分析(mass spectrometry, MS)等方法,操作简单、易于自动化,但仪器昂贵,在实验室难以普及[5]。而且,其结果都必须由测序法确定SNP的位置及突变类型[7]。
等位基因特异性扩增法检测较RFLP、SSCP快捷而廉价,但其实验条件要求较高,需严格控制,否则易出现假阳性[5]。为进一步降低成本与提高检测的特异性,本研究在等位基因特异性扩增法的基础上,以检测子宫内膜癌ERα基因富含GC区SNP 729C>T为例,建立了一种更为简便、准确而廉价的检测富含GC区的SNP方法。
1 材料与方法
1.1 研究对象将2000年6月-2007年2月在西安交通大学第一附属医院、空军军医大学西京医院、陕西省人民医院、陕西省肿瘤医院以及西安市第四人民医院妇科接受根治性子宫或全子宫切除手术的22例原发性子宫内膜癌患者作为病例组,年龄30~80岁,采集病变组织标本。选取同期接受手术的良性疾病患者42例作为对照组,年龄31~79岁,采集无病变组织标本。所有标本常规液氮罐保存。所有患者排除肝肾疾病,取材之前未接受放疗、化疗及任何激素治疗。
1.2 材料与试剂
1.2.1实验试剂 DNA Marker DL 2000[宝生物工程(大连)有限公司]、50bp DNA Ladder(TIAGEN生物科技有限公司)、DNA提取试剂盒(临床样品基因组DNA小量抽提试剂盒SK1341,生工生物工程技术服务有限公司)、PCR反应试剂盒(2×TaqPCR Master Mix,2×Pfu PCR Master Mix,2×HortstartTaqPCR Master Mix,TIAGEN生物科技有限公司)、PCR产物纯化与测序试剂盒(由北京奥克生物技术有限责任公司购自ABI公司)、溴化乙锭与溴酚蓝(Sigma),琼脂糖凝胶(Biowest公司,上海YITO 公司分装)、Tris(Amresco)。
1.2.2主要仪器与分析软件 液氮冻存装置(DeltaRange IC50R)、核酸蛋白分析仪(Bio-RAD SmartSpec Plus USA)、紫外线凝胶成像分析仪(SYNGENE Bio Imaging System USA)、PCR扩增仪(PERKIN ELMER GeneAmp Pro System 2400)、基因测序仪(北京奥克生物有限公司购自ABI-PRIMTM3730 XL)以及电泳仪(美国BIO-RAD公司);CHROMAS软件、BLAST(bl2seq)软件。
1.3 组织DNA提取按照生工生物工程技术服务有限公司临床样品基因组DNA小量抽提试剂盒说明严格进行操作。
1.4 技术原理等位基因特异性引物PCR(ASP-PCR)技术原理:利用单核苷酸多态性的等位基因位点特异性设计引物,即设计其3′端碱基与等位基因位点相应碱基相同的、与等位基因位点上游(5′端)的一小段DNA序列相同的引物即上游引物;或者是设计其3′端碱基与等位基因位点相应碱基互补的、并与等位基因位点下游(3′端)的一小段DNA序列互补的引物即下游引物,作为等位基因特异性引物。
1.5 技术方法的建立将两对引物分别加入单管中进行PCR扩增,所设计的两条特异性引物即内引物分别与DNA双链的两条单链相同,其3′端正好与SNP位点重合,由引物的3′端控制着引物的延伸反应,根据延伸产物的条带确定SNP类型;并通过在引物的3′端区域倒数第3位引入一个人为不匹配碱基来提高延伸反应的特异性[5]。具体原理见图1和表1。P1和P2为扩增含ERα Exon3 SNP位点C729T片段的外引物,产物为150 bp,无特异性,为对照扩增产物;S1和S2为SNP特异性引物;S3和S4也是SNP特异性引物,除3′端倒数第3个碱基突变外,其余序列与S1和S2相同。S1/S3与P2可引导C基因型扩增,产物为70bp;S2/S4与P2可引导T基因型扩增,产物为70 bp;由于3′端的特殊设计和严格的扩增条件,S1/S3和S2/S4决定扩增产物的特异性。3种扩增产物在体积分数为10 mL/L的琼脂糖电泳时易于分离、明确识别。
1.6 引物设计与合成依据SS-PCR原理,参照SASAKI等[8]方法,通过网站http://ncbi.nlm.nih.gov/,在GenBank获取ERα Exon3序列,设计扩增SNP位点C729T的多对引物,由北京奥科生物技术有限公司合成,其序列见表1。SNP位点C729T Exon位置:Exon3;mRNA位置:1021 C/T;DNA位置:AF123495 Exon 3-C237T;突变效果:Silent。
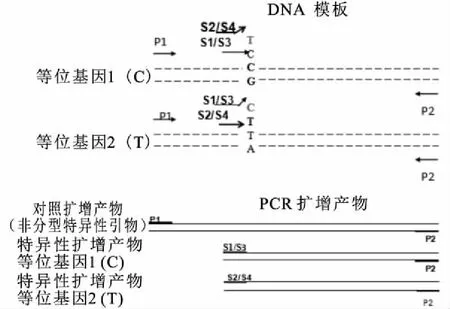
图1 引物特异性选择扩增分析ERα Exon3 C729T方法示意图
Fig.1 Analysis of C729T of Exon 3 in ERα by sequence-specific polymerase chain reaction (SS-PCR)
1.7 富含GC区序列特异性聚合酶链反应(SSP-PCR)的建立
1.7.1第1次PCR 以第一对引物P1与P2将22例子宫内膜癌与42例对照的所有DNA进行第1次PCR,扩增ERα基因的Exon3得到第1片段。按PCR反应试剂盒说明进行操作。
于冰上在0.2 mL灭菌离心管依次加入模板DNA 1~5 μL,P1、P2(10 μmol/L)各1.0 μL、2×Pfu PCR Master Mix 12.5 μL,补ddH2O至25 μL。反应参数:预变性94 ℃,3 min;变性94 ℃,60 s,退火温度57 ℃(预试验优化:由59 ℃开始,每次递降1 ℃,直到54 ℃),60 s,延伸72 ℃,60 s,循环30次;72 ℃延伸8 min。20 g/L琼脂糖凝胶电泳及成像分析。
表1 扩增ERα基因Exon3片段的引物序列
Tab.1 Amplified primer sequence of Exon3 fragment in ERα gene
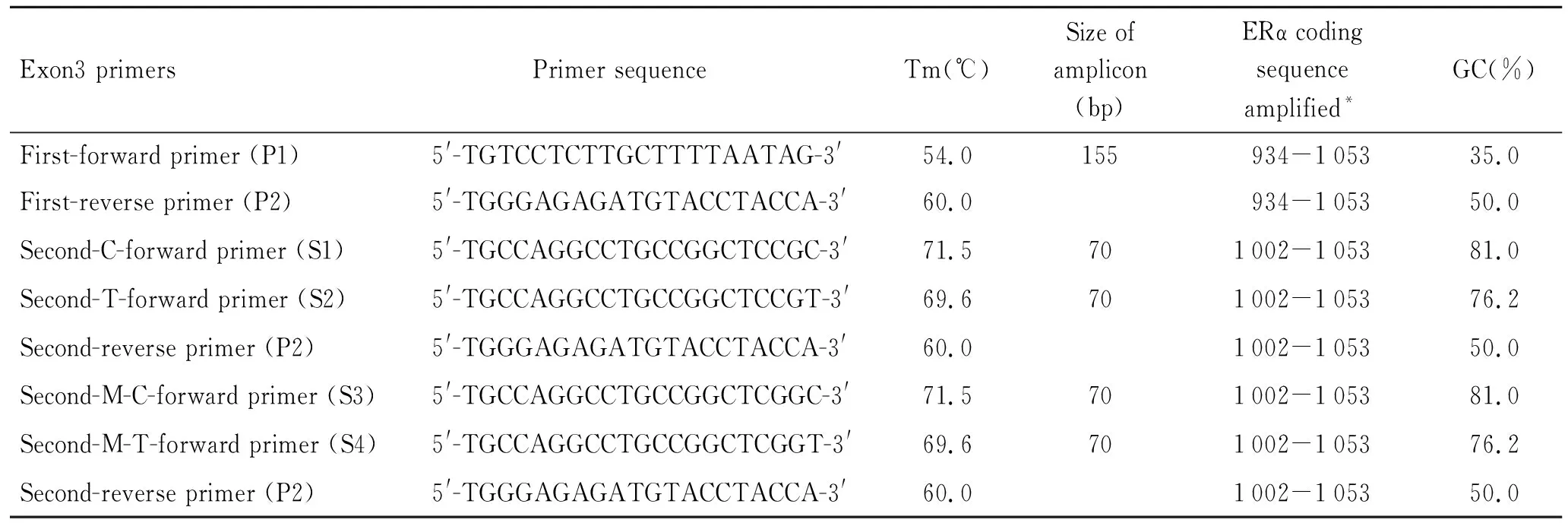
Exon3 primers Primer sequenceTm(℃)Size ofamplicon(bp)ERα codingsequenceamplified*GC(%)First-forward primer (P1)5'-TGTCCTCTTGCTTTTAATAG-3'54.0155934-105335.0First-reverse primer (P2)5'-TGGGAGAGATGTACCTACCA-3'60.0934-105350.0Second-C-forward primer (S1)5'-TGCCAGGCCTGCCGGCTCCGC-3'71.5701002-105381.0Second-T-forward primer (S2)5'-TGCCAGGCCTGCCGGCTCCGT-3'69.6701002-105376.2Second-reverse primer (P2)5'-TGGGAGAGATGTACCTACCA-3'60.01002-105350.0Second-M-C-forward primer (S3)5'-TGCCAGGCCTGCCGGCTCGGC-3'71.5701002-105381.0Second-M-T-forward primer (S4)5'-TGCCAGGCCTGCCGGCTCGGT-3'69.6701002-105376.2Second-reverse primer (P2)5'-TGGGAGAGATGTACCTACCA-3'60.01002-105350.0
*ERα 编码序列参见Genbank M12674。引物序列设计参见SASAKI等方法[8]。
1.7.2测序 随机从第1次PCR产物中抽取10例,PCR产物送北京奥科公司按试剂盒操作指南进行PCR产物纯化,以第一对引物的上游引物、按照染料终止子循环测序试剂盒说明应用ABI-PRIMTM3730 XL测序。结果参照GenBank中ERα Exon3编码序列(AF123495),用CHROMAS软件读取测序彩图,将所得DNA序列输入计算机后,利用[http://www.ncbi.nlm.nih.gov/]在线BLAST(bl2seq)软件对位排列,并辅以人工校对。
1.7.3第2次PCR 以引物S1/S3与P1,S2/S4与P2,以已扩增的第1片段为模板,按PCR反应试剂盒(2×Pfu PCR Master Mix/2×TaqPCR Master Mix/2×Hortstart PCR Master Mix)按说明进行操作,扩增ERα基因的Exon3第2片段。
于冰上在0.2 mL灭菌离心管依次加入模板(第1片段DNA稀释20倍)1 μL、S1/S2或S3/S4(10 μmol/L)均0.25 μL、P2(10 μmol/L)0.5 μL、2×Pfu PCR Master Mix/2×TaqMaster Mix/2×Hortstart PCR MasterMix 6~6.25 μL,补ddH2O至12.5 μL。反应参数:预变性94 ℃,3 min;变性94 ℃,30 s,退火85 ℃/69.5 ℃/68 ℃,30 s,延伸72 ℃,30 s,循环30次;最后72 ℃延伸7 min。20 g/L琼脂糖凝胶电泳及成像分析。
进一步优化反应条件,将模板、引物浓度以及2×PCR MasterMix的条件改变一个,其余固定,分别优化反应条件。模板即第1片段DNA:取其原液、10倍稀释液、20倍稀释液各1 μL;引物S1、S2与P2(10 μmol/L)分别为0.125、0.25、0.5 μL;2×PCR Master Mix分别为3.125 μL、6 μL、6.25 μL进行扩增。得知最适条件:第1片段DNA 20倍稀释1 μL;引物S1、S2与P2各0.25 μL;2×PCR Master Mix 6~6.25 μL。
1.8 富含GC区序列特异性PCR(SSP-PCR)的应用——序列特异性PCR用于鉴别Exon3 SNP位点C729T基因型
1.8.1第2次PCR 以优化的引物S3与P2、S4与P2和2×HortstartTaqPCR Master Mix,将22例子宫内膜癌与42例对照的第1次PCR产物作为模板,进行第2次PCR。按照试剂盒说明书操作,反应体系总体积12.5 μL,PCR反应参数的退火温度68 ℃,具体同前。
1.8.2直接测序法检验基因型 将上述所有样本第1次扩增产物(子宫内膜癌22例,对照42例)送奥科公司,用第1对引物的上游引物,按试剂盒操作指南进行PCR产物纯化、DNA序列测定及分析。操作同前。
2 结 果
2.1 第1次PCR扩增结果22例子宫内膜癌与42例对照均进行第一次PCR,20 mL/L琼脂糖凝胶电泳第1次PCR产物成像分析结果,产物大小均为155 bp。
2.2 第2次PCR扩增结果对已测序示C/C纯合型的10例,用引物S1、S2与P2和2×Pfu PCR Master Mix进行第2次PCR扩增,结果显示:当退火温度为60 ℃,始终扩增出S1P2(C)、S2P2(T)片段,均为70 bp,呈C/T杂合型(图2)。
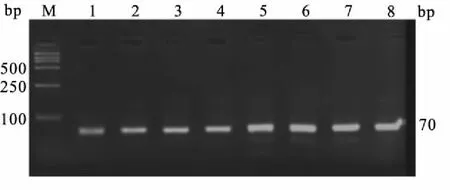
图2 用引物S1、S2与P2和Pfu DNA聚合酶第2次PCR的结果
Fig.2 The results of gel electrophoresis of the second PCR products amplified with 3′base specific primers S1, S2 and Pfu DNA polymerase
M:DNA Marker DL 2000;奇数孔:4个样本的C扩增片段,偶数孔:4个样本的G扩增片段。
对测序示C/C纯合型的10例,用引物S1、S2与P2和2×TaqPCR Master Mix进行第2次PCR扩增,结果显示退火温度49 ℃至85 ℃,于83 ℃以前,始终能扩增出S1P2、S2P2片段,即C/T杂合型,其中80 ℃至83 ℃,S2P2片段(T)较S1P2(C)片段亮度减低;直至85 ℃时,才仅扩增出S1P2片段,即显示C/C野生型(图3A、图3B、图3C)。
对测序示C/C纯合型的10例,用引物S3与P2、S4与P2和2×TaqPCR Master Mix进行第2次PCR扩增,退火温度依次为60 ℃,62 ℃,69 ℃,69.5 ℃,70 ℃,于退火温度69.5 ℃之前,显示C/T杂合型;而于69.5 ℃,结果显示与测序显示C/C野生型一致。可见,随着退火温度的升高,T较C渐淡,特异性渐提高。而70 ℃时C和T均未扩增(图4A、图4B)。
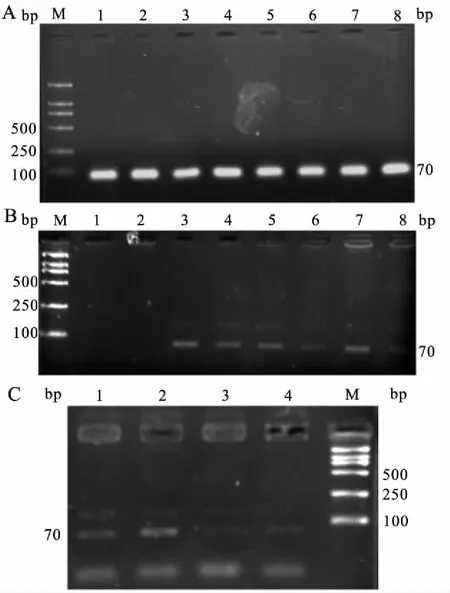
图3 用引物S1、S2与P2和Taq DNA聚合酶及不同退火温度的PCR结果
Fig.3 The results of gel electrophoresis of the second PCR products amplified with 3′base specific primers and Taq DNA polymerase in various Tm
M:DNA Marker DL 2000。A,1、2:49 ℃;3、4:59 ℃;5、6:69 ℃;7、8:77 ℃;奇数孔:S1P2片段(C);偶数孔:S2P2片段(T)。B,1、2:不加引物S1、S2的阴性对照;3、4:79 ℃;5、6:80 ℃;7、8:81 ℃;奇数孔:S1P2片段(C);偶数孔:S2P2片段(T)。C,1、2:82 ℃;3、4:83 ℃;奇数孔:S2P2片段(T) ;偶数孔:S1P2片段(C)。
对测序示C/C纯合型的10例,用引物S3、S4与P2和2×HortstartTaqPCR Master Mix,进行第2次PCR扩增,退火温度分别为60 ℃、65 ℃、66 ℃、67 ℃、68 ℃,于退火温度68 ℃之前,均为C/T杂合型,但65 ℃时,S2P2片段(T)较S1P2(C)片段亮度减低;于68 ℃,S2P2片段(T)消逝,仅显示S1P2(C)片段,即C/C纯合型,与测序结果一致(图5A、B)。
2.3 序列特异性PCR用于鉴别Exon3 SNP位点C729T基因型结果
2.3.1第2次PCR 以所有子宫内膜癌与对照的第1次PCR产物作为模板,应用优化的引物S3、S4与P2和2×HortstartTaqPCR Master Mix,进行第2次PCR扩增,结果显示,始终扩增出S3P2片段,即C/C野生型,扩增片段为70 bp。说明未发现Exon3位点C729T SNP(图6)。
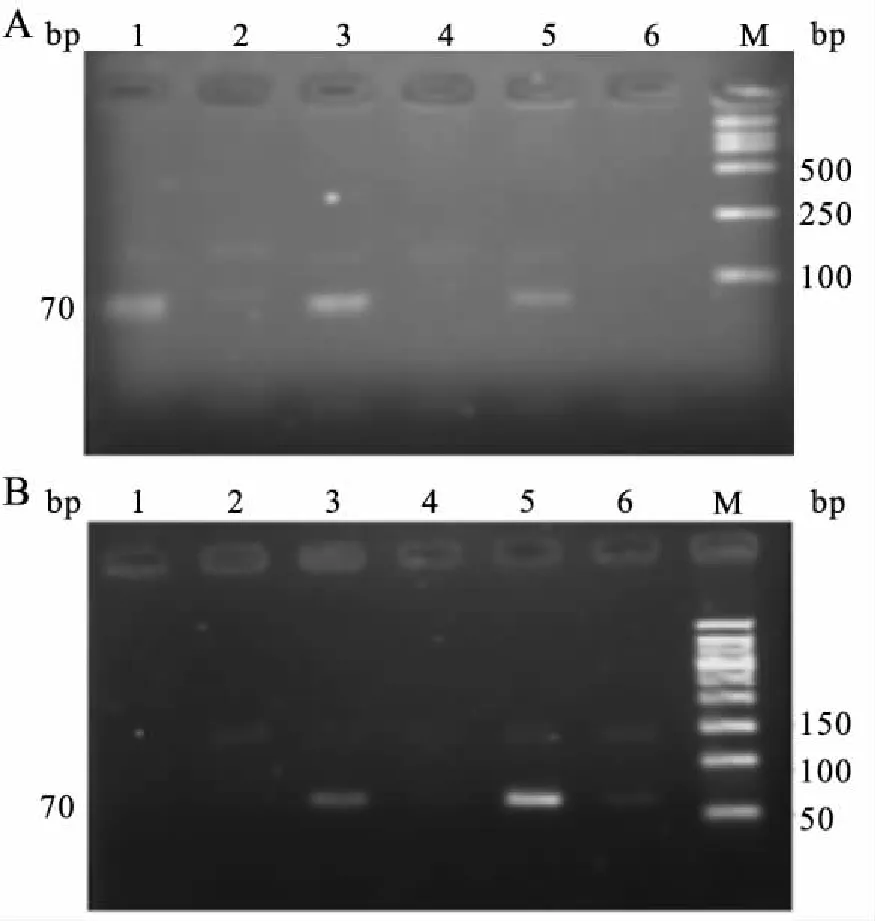
图4 用引物S3、S4与P2和TaqDNA聚合酶及不同退火温度的PCR结果
Fig.4 The gel electrophoresis result of the second PCR products amplified with 3′ base specific primer with a mismatch at the third 3′-terminal base andTaqDNA polymerase at various Tm
A,1、2:67 ℃;3、4:68 ℃;5、6:69.5 ℃;奇数孔:S3P2片段(C);偶数孔:S4P2片段(T);M:DNA Marker DL 2000。B,1、2:70 ℃;3、4:69.5 ℃;5、6:69 ℃;奇数孔:S3P2片段(C);偶数孔:S4P2片段(T);M:50 bp DNA Ladder。
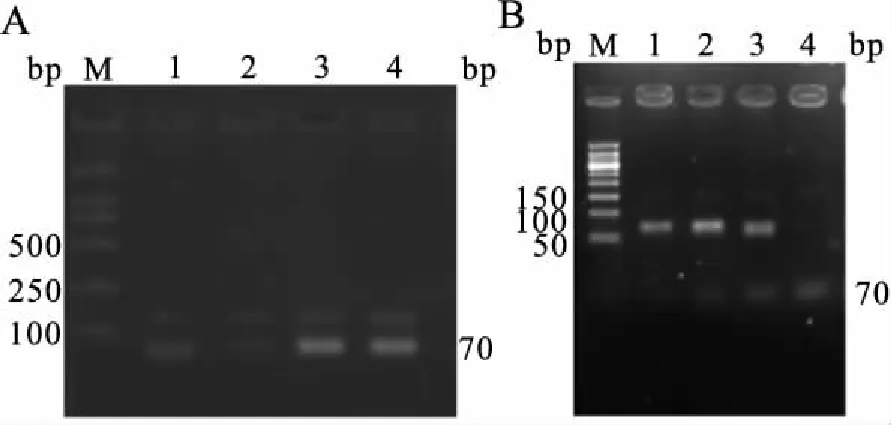
图5 用引物S3、S4与P2和Hortstart DNA聚合酶PCR的结果
Fig.5 The gel electrophoresis of the second PCR products amplified with 3′ base specific primer with a mismatch at the third 3′-terminal base and Hortstart DNA polymerase at various Tm
A, M: DNA Marker DL 2000;1、2:65 ℃;3、4:60 ℃;奇数孔:S3P2片段(C);偶数孔:S4P2片段(T)。B,M: 50 bp DNA Ladder;1、2:67 ℃;3、4:68 ℃;奇数孔:S3P2片段(C);偶数孔:S4P2片段(T)。
2.3.2直接测序法验证基因型 所有样本第1次扩增产物(子宫内膜癌22,对照42例)送奥科公司,用第1对引物的上游引物、按试剂盒操作指南进行PCR产物纯化、DNA序列测定及分析。均显示位点C729T基因型为C/C型。
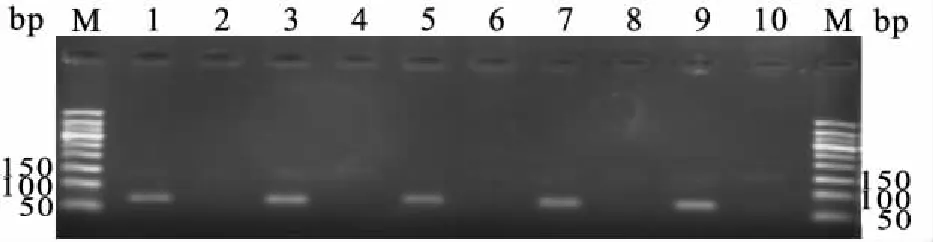
图6 序列特异性PCR鉴别Exon3 SNP位点C729T基因型结果
Fig.6 The gel electrophoresis of SS-PCR products amplified with 3′ base specific primer with a mismatch at the third 3′-terminal base and Hortstart DNA polymerase at 68 ℃
M: 50 bp DNA Ladder;奇数孔:S3P2片段(C);偶数孔:S4P2片段(T)。
3 讨 论
等位基因特异性扩增法是检测点突变较快捷的一种方法[6],但其实验条件要求较高,需严格控制, 否则易出现假阳性[5,9-10]。因此,本研究在等位基因特异性扩增法的基础上研究建立了一种准确、快速而廉价的SNP检测方法[ 9-10,11-14],并针对富含GC区的ERα基因SNP 729C>T扩增进行了方法学的探讨。
等位基因特异性PCR(ASPCR)主要依赖引物的设计及优化PCR反应条件,使得只有完全互补的序列才能扩增。如果错配位于引物3′末端导致PCR不能延伸,则叫四引物扩增阻滞突变系统(amplification refractory mutation system, ARMS)[5,9,15]。本研究建立SS-PCR与四引物扩增阻滞突变系统PCR(ARMS-PCR)相似,与文献报道一致[9,11-14];与直接测序结果进行对照,检测准确率达100%,说明其检测结果准确。
3.1 3′端碱基特异性聚合酶链反应(3′base specific polymerase chain reaction, 3′BS-PCR)的特异性当采用仅3′末端一个碱基特异的引物S1和S2测定SNP类型时,于85 ℃前检测始终出现两条条带,即杂合型C/T,与测序结果显示的野生纯合型C/C不符(图2、图3)。考虑原因是该SNP处于富含GC区,仅3′端一个碱基特异的引物与DNA模板退火时,不足以影响该引物的延伸反应。提示3′BS-PCR对富含GC区的SNP分型的特异性有限。该结果与汲振余等[16]相似,其在温度升至61 ℃前对测序预先检测为单纯变异株的感染却扩增为两条带(显示变异株与野生株都有的混合感染)。不同的是,其检测的突变点并非在富GC区,所以在低于引物Tm值3 ℃时即选择出合适退火温度,本课题组经过相当长时间摸索才达到与测序一致的结果,而且适合的退火温度高达85 ℃。值得一提的是,这与SASAKI等[8]的结果有极大差异,其实验的退火温度为60 ℃,但我们却始终不能重复其实验结果,考虑该区富含C、G(达81.0%、76.2%)所致。也正因为此,本研究重新设计该技术方案并进行探讨。
3.2 引入3′端人工错配碱基提高SNP分析特异性当采用引物S3、S4,也就是在一对3′端特异性引物S1、S2的3′端区域倒数第3个位点加入1个人工错配碱基(与模板相同)时,模板CC仅出现一条与等位基因C相一致的电泳条带,进一步说明特异性引物的3′端在与DNA 模板退火时,仅3′端一个碱基的不同不足以影响该引物的延伸反应;当3′端区域有两个与模板不互补的碱基时,可有效地阻止非特异性延伸反应。证明引入3′端人工错配碱基能有效提高富含GC区的SNP分型的特异性,这一结果与其他研究者的结果相似[12]。同理,如果模板是TT,则仅出现一条与等位基因T相一致的电泳条带。对杂合子模板CT来说,人工突变碱基的引入不影响检测结果而扩增出C和T两条带。遗憾的是,本研究样本没有检出杂合型C/T和突变纯合型T/T,这被测序结果证实(图4、图5),因为不存在杂合型C/T和突变纯合型T/T。
3.3 不同的聚合酶对特异性扩增反应的影响在方法建立之初,本研究应用Pfu DNA聚合酶扩增。由于该酶有3′端外切酶活性,虽然具有矫正在PCR时出现的碱基错配,但恰巧可能使3′端特异性引物相差的一个碱基被切除而失去其特异性,故始终扩增出两条带,显示杂合子C/T(图2)。随后,为了采用特异性引物的3′端之开关控制延伸反应的作用,本研究使用无3′端外切酶活性的TaqDNA聚合酶。目前该酶主要有两大类,即冷启动酶和热启动酶。本研究选择TiangenTaqDNA聚合酶和HotstartTaqDNA聚合酶,对这两类无3′端外切酶活性的Taq酶作了对比实验。同样采用3′端区域倒数第3个位点加入1个人工错配碱基的特异性引物及同样的酶浓度,发现TaqDNA聚合酶于退火温度为69.5 ℃(图4),而HotstartTaqDNA聚合酶在退火温度为68 ℃(图5)时,所得结果与测序结果一致。说明二者相差不大。但是,HotstartTaqDNA聚合酶价格较贵,使测定成本增加。提示不同的聚合酶对特异性扩增反应的影响不同[15]。
3.4 退火温度对特异性扩增反应的影响退火温度与引物的Tm值关系密切,因此,选择合适的退火温度非常重要[17]。针对富含GC区的ERα基因中729突变点来说,所设计的两条错配特异性引物适宜的Tm值为69~70 ℃。当退火温度为68 ℃或69.5 ℃时(图4、图5),可得到较高的特异性扩增产物量和较好的扩增特异性。所以,退火温度选择为比特异性引物的Tm 值低1~3 ℃为宜。
3.5 特异性引物浓度对特异性扩增产物的影响为了提高扩增的特异性,以CC为模板,设计了一系列不同浓度的引物进行实验比较。在12.5 μL体系中,外引物P1和P2浓度固定为10 μmol/L(10 μmol/L),量为0.5 μL,两个特异性引物S1、S2或S3、S4在10 μmol/L的量依次为0、1.25、0.25、0.5 μL。结果表明,当0.25 μL时既提高了特异性扩增,也保证了特异性带的强度。
3.6 酶浓度的选择酶的浓度直接影响到扩增的质量和产量。酶浓度过高,非特异性扩增带会增多;酶浓度过低,则会降低靶序列的扩增量。一般来说,在其他PCR条件都调整至最佳时,每100 μL PCR反应体系中含酶1~2.5 U为宜,但酶的用量仍需根据不同的模板分子或引物进行适当调整[15]。在本实验12.5 μL的PCR 反应体系中,0.6 U或0.625 U的酶用量即可清晰地判断SNP类型。
3.7 其它因素靶DNA的浓度也很重要。第2次PCR以第1次PCR产物为模板时,如果浓度太高,体系中会有过量的总Mix,不仅电泳时可显示第1片段,而且可能影响第2片段特异性扩增,出现非特异性片段。我们经过选择将第一片段原液稀释20倍,12.5 μL体系取1 μL为宜。另外,在扩增实验中,dNTP浓度、Mg2+浓度与酶的活性相关[15]。本实验用的是总Mix,其量主要靠经验摸索。
3.8 与直接测序法在SNPs鉴定的比较本研究所建立的方法操作简单、周期短、费用低廉,其可靠性进一步被测序法所证实。
检测SNPs的经典方法是RFLP-PCR、SSCP-PCR等[5],其结果仅能判断SNPs的有无,而无法确定多态位点的碱基类型,需要测序方法最终验证[7]。测序法检测SNP的效率达到100%,但测序检测SNP的方法花费昂贵。故对一些比较小、外显子相对较少的基因的SNP鉴定可以直接测序,而对一些较大的、外显子较多的基因不宜用测序法直接检测SNP,也不适用于临床对大量的标本进行检测[5]。
本研究扩增Exon3位点C729T的片段为155 bp,由于扩增片段短,故对所有样本采用PCR产物直接测序法,以验证所建立的SS-PCR的准确性。结果与大量文献报道的相似,成功率接近100%[7]。因此,我们认为,直接测序是当前检测SNP的最可靠的方法。但也存在费用偏高、过程较繁琐及周期较长等不足。相信随着测序技术的不断成熟和测序成本的降低,直接测序将会大规模地用于SNP的检测与分型[7]。
3.9 评价与展望3′BS-PCR法操作简单,周期短,准确,费用低廉,适合于检测已知位点的定点突变,特别适合于大规模筛查。关键是应固定所用试剂及仪器和把握最佳退火温度。因而应在引物、DNA模板浓度、dNTP、Taq聚合酶等严格控制情况下,以测序法证实的已知一定数量的纯合、突变样本,摸索最佳退火温度及退火时间[15]。应特别注意引物G+C百分含量决定引物Tm,影响退火温度,因而不同实验条件下退火温度和时间亦应不同。只要严格掌握最佳实验条件,本法对于测定点突变具有较好的应用价值[10,12,14]。
本研究证明了等位基因专一性的引物的3′端第3位碱基引入不配对可以降低引物的非特异性延伸。而且,与3′BS-PCR在适合的退火温度上有着巨大的差异。因此,基于等位基因专一性PCR的实验方法均可以考虑通过3′端第3位碱基引入不配对来降低非特异性延伸,减少实验条件优化的时间。
本文建立的方法在特异性引物的3′端区域人为引入一个不匹配碱基,提高了扩增特异性,降低了假阳性率。该方法实现了检测3种基因型,电泳即可分析,不需要大型仪器,具有快速、简便、成本低等优点[5,12];目前反应体积为12.5μL,如将PCR反应体系减小到数微升,选用96或384孔板对多个样品同时操作,则可大大提高SNP检测效率;本方法可用于制备基因快速检测试剂盒在基层医疗机构推广使用[11-12]。但该方法仅适于已知的突变;由于该方法对引物设计要求较高,且涉及多重PCR,优化过程较为繁琐,条件较严格,难以实现高通量自动化检测。未来若采用自动化的并行液体加入装置,将更节省时间。
4 结 论
本研究通过优化序列特异PCR检测SNP的条件,建立了适合富含GC区SNP分型的序列特异性PCR方法,成功用于子宫内膜癌特定ERα Exon3位点C729T多态性基因分型,有望推广应用。
