噻吩、吡咯、呋喃在H-FAU分子筛中吸附和扩散行为的分子模拟
2019-10-19杨晓东刘熠斌杨朝合
党 宇,杨晓东,刘熠斌,冯 翔,杨朝合
(中国石油大学 重质油国家重点实验室,山东 青岛 266580)
八面沸石(FAU)分子筛因具有较高的内表面积、适宜的孔结构、固体酸特性及高温稳定性而被用作催化裂化(FCC)反应的催化剂[1-2]。目前,催化裂化过程正逐步适应加工劣质的重油,而劣质重油中含有大量杂原子化合物。杂原子化合物通过诱导效应来中和催化剂的酸中心,或以形成焦炭覆盖酸中心的方式使催化剂中毒失活,严重阻碍其它烃类的反应[3-4]。
原料分子中的杂原子主要包括S、N、O原子,不同的杂原子化合物对催化裂化反应的影响程度不同。研究表明[5],杂原子具有给电子共轭效应且分散正电荷能力强,因此极易在酸作用下质子化,引发开环或聚合反应。与烃类分子相比,杂原子化合物在分子筛上吸附能力强,扩散性能差。考察典型杂原子分子在分子筛上的吸附和扩散行为,对研究劣质重油非理想组分阻滞原料转化的作用机理有重要意义。
分子模拟方法已被广泛用于研究反应物分子在多孔材料中的吸附和扩散性能[6-8]。该方法能够准确的计算客体分子和分子筛之间的相互作用,得到客体分子的宏观、微观性能数据,如吸附等温线、吸附热和分子在孔道中分布等。目前,众多学者采用分子模拟方法计算了杂原子分子在FAU分子筛中的吸附行为,得到了与实验一致的结果。沈喜周等[9]采用蒙特卡洛方法(GCMC)考察了噻吩和吡啶在FAU分子筛上的吸附行为,计算得到的吸附热数据和实验结果一致;同时,研究发现噻吩与分子筛孔道的相互作用强于吡啶。杨文平等[10]采用GCMC方法模拟了噻吩类硫化物在FAU分子筛中的吸附性能,结果表明,噻吩、2-甲基噻吩和2,5-二甲基噻吩在FAU上的单组分吸附等温线均可用Langmuir吸附模型拟合;此外还发现,在573.15 K、0.01~1.0 MPa下,3种分子在FAU孔道中存在竞争吸附,对含硫化合物在催化反应中的阻滞作用机理提供了微观解释。Fu等[11]通过GCMC方法模拟计算了苯和噻吩在FAU分子筛中的竞争吸附,结果表明,2种分子在FAU超笼中的吸附位随吸附量的变化而改变;分子筛酸性质(硅/铝比)对2种分子的吸附能和分布有较大影响。
大量工业数据表明,氮化物较强的吸附性会严重影响催化剂的活性,给反应过程带来不利影响[12-16]。部分学者模拟了氮化物在FAU分子筛中的吸附行为。熊袖章等[17]采用GCMC方法考察了吡啶衍生物在FAU中的吸附行为,得到了吸附等温线、吸附平衡常数和吸附热数据,与实验结果一致,并发现不同客体分子的吸附性能与其碱性和结构有关。Injan等[18]通过DFT方法计算了吡啶在H-FAU分子筛团簇上的吸附,得到了和实验一致的吸附热结果。王迪等[19]考察了吲哚对减压蜡油(VGO)在FAU催化剂上裂化反应的影响,结果表明,随着原料中氮含量的增加,反应转化率明显降低、干气选择性变差、汽油和液化气收率显著下降;分析认为是由于吲哚在反应中优先吸附导致催化剂活性中心减少所致。
有机含氧化合物会导致原油的化学性质不稳定,降低油品质量,因此氧化物在分子筛中的吸附特性对催化剂性能的影响同样重要,但关于油品中氧化物在FAU分子筛的吸附行为的研究还未见报道。同时,有关杂原子化合物在FAU分子筛孔道中的扩散行为的研究仍存在空白;对S、N、O这3种杂原子化合物在催化剂中吸附和扩散性能的定量对比研究还未见报道。
笔者采用GCMC方法和分子动力学(MD)方法,在催化裂化温度(823 K)下,考察了劣质重油中典型的五元环杂原子化合物噻吩、吡咯和呋喃在H-FAU分子筛中的吸附和扩散性能,得到了它们的吸附等温线、吸附热、吸附质分布及其在孔道中的扩散系数。模拟结果为催化裂化过程中不同类型杂原子分子对反应影响的研究提供理论指导。
1 计算模型与方法
1.1 分子筛模型
FAU分子筛属于立方晶系,空间群为Fd-3m,晶胞参数a=b=c=2.503 nm,α=β=γ=90°,主要结构是八面沸石笼,笼口孔径为0.74 nm×0.74 nm。模拟过程中,按文献[20-21]调整分子筛硅/铝原子比为5.86,与实际使用分子筛的硅/铝比一致,并引入H原子平衡电荷,构建H-FAU分子筛模型。初始H-FAU分子筛模型的分子式为:H28O384Al28Si164,其中各原子电荷指定如下[20-21]:Si(+0.89)、Al(+0.73)、O(-0.33)、H(+0.083)。模拟中使用1(1×1×1)个单元晶胞,并在x,y,z3个方向添加周期性条件。对更大晶胞进行测试未发现明显的尺寸效应,表明以上晶胞大小合适。最终的 H-FAU 模型和孔道特征如图1所示。
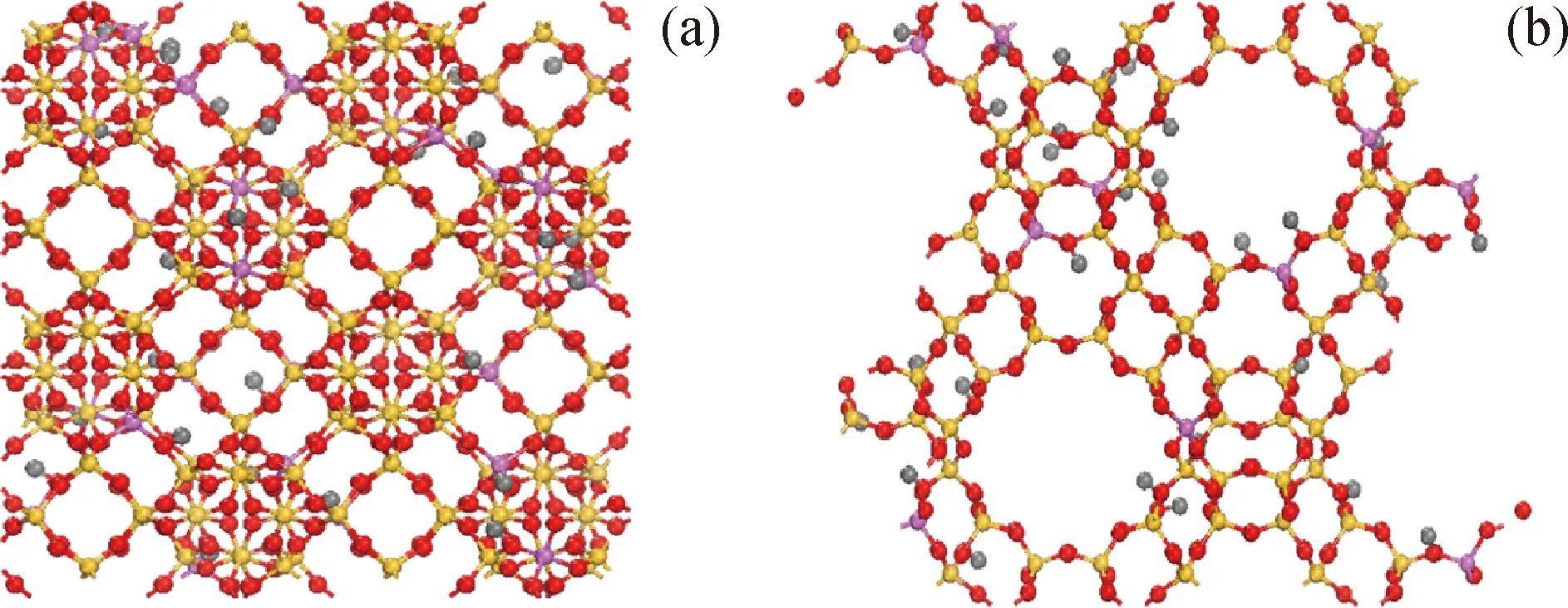
图1 H-FAU分子筛模型及其孔道特征Fig.1 Model and pore structure of H-FAU zeolite(a)H-FAU zeolite model;(b)Pore structure of H-FAU zeoliteAl—Pink;Si—Yellow;O—Red;H—Gray
1.2 杂原子化合物模型
噻吩、吡咯和呋喃3种化合物的三维尺寸如表1 所示。由表1可知,3种分子的最小截面尺寸分别为:噻吩0.55 nm×0.21 nm、吡咯0.55 nm×0.22 nm、呋喃0.51 nm×0.20 nm,均小于FAU分子筛超笼笼口孔径0.74 nm×0.74 nm。因此,理论上3种分子可以扩散进入FAU分子筛孔道中。采用分子模拟软件Dmol3模块对3种分子进行结构优化,并计算了各原子的ESP电荷,计算中使用了广义梯度近似(GGA)的Perdew-Burke-Ernzerhof(PBE)泛函和双数值轨道基组(DNP)[22-23],结果如图2所示。

表1 3种杂原子化合物分子的三维尺寸Table 1 The size of three heteroatom molecules

图2 噻吩、吡咯、呋喃的结构示意图以及计算ESP电荷Fig.2 Molecular structures and calculated ESP charges of heteroatom molecules(a)Thiophene;(b)Pyrrole;(c)FuranH—White;C—Gray;S—Yellow;N—Blue;O—Red
1.3 参数设置
模拟软件Sorption模块中抽样方法选用Metropolis方法,客体分子和分子筛的相互作用包括静电作用和范德华作用,其中范德华作用采用Lennard-Jones势函数,数学表达式为:
(1)
式(1)中,ULJ表示客体分子和分子筛之间相互作用;i和j表示不同原子;Rij表示原子间距,nm;Dij和(R0)ij为Lennard-Jones参数;qi和qj表示原子所带电荷。模拟中采用Universal力场,静电相互作用采取Ewald方法处理,非键相互作用的加和方法采用Atom based算法,范德华作用截断距离设置为1.251 nm,正好小于晶胞边长(2.503 nm)的一半。计算平衡步数为106步,生产步数为107步。
由于缺乏吡咯和呋喃的吸附热数据,但噻吩和吡啶的吸附热结果已有详细数据。因此,为了验证模拟参数的合理性,首先采用上述模拟参数计算了给定实验条件[24-25]下噻吩和吡啶在FAU分子筛中的Henry常数,并采用范特霍夫(van’t Hoff)方程计算吸附热:
Qst=RT2(∂lnk/∂T)0
(2)
式(2)中,Qst为吸附热,kJ/mol;R为气体常数,J/(mol·K);k为Henry常数,T为反应温度,K。计算得到噻吩和吡啶在FAU上的吸附热分别为93.45 kJ/mol和59.75 kJ/mol,与实验值80.22~94.08 kJ/mol(噻吩)[25]和57.32~98.74 kJ/mol(吡啶)[24]一致。这表明上述模拟参数能够准确的描述杂原子化合物和分子筛的相互作用。
动力学性质采用Forcite模块计算,该模块主要依据分子动力学(MD)方法,MD模拟中采用NVE系综,温度为FCC反应温度823 K。牛顿运动方程的积分采用蛙跳算法(Velocity-Verlet),模拟步长1 fs,总模拟时长1 ns,其中前500 ps用于平衡体系。扩散系数Ds采用爱因斯坦方程计算:
(3)
式(3)中,Ds表示扩散系数,m2/s;N表示单位晶胞内分子数;r(0)是客体分子质心的初始位置坐标,r(t)是客体分子在时间t时的质心坐标,[|r(t)-r(0)2|]是扩散分子均方根位移(MSD)的系综平均。模拟中3种杂原子化合物在分子筛中的负载量均为每单元晶胞26个分子。
2 结果与讨论
2.1 吸附等温线分析
图3为823 K、100~1000 kPa条件下,噻吩、吡咯和呋喃的单组分在H-FAU中的吸附等温线。

图3 823 K下噻吩、吡咯和呋喃在H-FAU分子筛中的吸附等温线Fig.3 Adsorption isotherm of thiophene,pyrrole and furan in H-FAU zeolite at 823 K
从图3可以看到,在整个压力范围内3种分子的吸附量随压力的增大缓慢增加。低压下3种分子吸附量的增速依次为:噻吩、吡咯、呋喃,表明 H-FAU 分子筛对噻吩的吸附能力最强,对呋喃的吸附能力最弱。采用朗格缪尔(Langumir)吸附等温线模型(公式(4))对3种分子的吸附数据进行拟合,结果如表2所示。
(4)

表2 噻吩、吡咯和呋喃在FAU分子筛上吸附的Langmuir吸附模型拟合参数Table 2 Adsorption parameters of isotherms of thiophene,pyrrole and furan in FAU zeolite
式(4)中,N表示单位晶胞内分子数;Nm为单元晶胞饱和吸附分子数;B为平衡常数,kPa-1。
由图3和表2的结果可知,3种分子在FAU分子筛中的吸附数据能够较好的采用Langmuir吸附方程拟合,相关系数(R2)均大于0.99。此外,3种分子的吸附等温线属于Ⅰ型等温线,表明呈现典型的单分子层吸附。
表2中Langmuir方程拟合得到的3种分子的饱和吸附量大小顺序依次为:噻吩、吡咯、呋喃,且噻吩的饱和吸附量是吡咯的1.17倍,是呋喃的1.21倍。这进一步证明,与吡咯和呋喃相比,噻吩与FAU分子筛有更强的相互作用。
为了考察实际反应条件下3种杂原子化合物组分在催化剂中的实际吸附行为,进一步计算了 823 K 下3种组分的等摩尔比混合气体在分子筛中的吸附等温线,模拟结果如图4所示。由图4可知,混合气体中3种组分的吸附量随压力的变化趋势与单组分的吸附情况相似。值得注意的是,在低压范围内,噻吩的吸附量随压力的增大而迅速增加,而吡咯和呋喃的吸附量变化较为平缓。同时,在各自分压为700 kPa时3种分子的吸附均达到饱和,饱和吸附量的大小次序依次为噻吩、吡咯、呋喃,其中噻吩的饱和吸附量是吡咯的1.28倍,呋喃的1.18倍。这表明3种分子在H-FAU分子筛中存在比较明显的竞争吸附,其中噻吩的吸附能力最好,呋喃相对较弱。由于3种杂环分子的结构相似,均为五元环化合物,且计算的分子三维尺寸接近,因此吸附量和吸附变化趋势的差异主要是受杂原子性质的影响。这说明在823 K、100~1000 kPa条件下,杂环化合物对催化裂化反应的影响由大到小依次为噻吩、吡咯、呋喃。
2.2 吸附热分析
对于气体在多孔材料中的吸附过程,吸附热的其大小可用于定量描述客体分子和多孔材料之间相互作用大小。在GCMC模拟中,等量吸附热ΔQ可由巨正则系综中能量/粒子的涨落[26]计算得到:
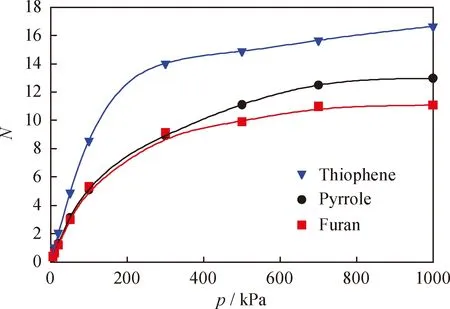
图4 823 K下,噻吩、吡咯和呋喃混合气体在H-FAU分子筛中吸附等温线Fig.4 Adsorption isotherm of thiophene,pyrrole and furan mixtures in H-FAU zeolite at 823 K
(5)
式(5)中,ΔQ为等量吸附热,kJ/mol;T为体系温度,K;R表示理想气体常数,J/(mol·K);UN为吸附相势能;〈〉为系综平均。823 K下,3种杂环分子在H-FAU分子筛中的吸附热曲线如图5所示。从图5中可以看到,噻吩和吡咯的吸附热均随压力的升高而增加。这说明:当吸附量较低时,客体分子和分子筛孔道之间的相互作用占主导地位;随着压力的增大(即吸附量的增加),客体分子之间的相互作用增强[27-28],导致吸附热增加。此外,从图5中还可以看到,低压下噻吩的吸附热随压力变化明显快于吡咯,说明噻吩的热效应更强。而呋喃的吸附热曲线表现出与噻吩和吡咯完全不同的变化趋势,其大小随压力的增大而降低。这与很多传统小分子气体的吸附呈相同的规律,表明在低压下H-FAU分子筛对呋喃分子的捕捉效率较高,使得呋喃分子优先占据活性位,放出较多热量。但是随着压力增大,孔道表面活性位被占据,呋喃的吸附转向其它普通的吸附位,导致吸附热下降。这表明在实际反应较高压力下,呋喃对分子筛的影响要小于噻吩和吡咯,这一点也可以由饱和吸附时3种分子的吸附热大小次序(噻吩、吡咯、呋喃)得到证明。

图5 噻吩、吡咯和呋喃在H-FAU分子筛中的等量吸附热Fig.5 Isoteric heat of heteroatom molecules in H-FAU zeolite
2.3 吸附质分布
蒙特卡洛模拟可以得到一系列吸附质分子的构型数据,分析这些数据可以得到客体分子在分子筛孔道中各个吸附位点的出现概率,从而有助于我们进一步认识不同杂原子化合物在H-FAU分子筛中的分布情况。
823 K时,3种杂环化合物单组分在H-FAU分子筛中概率密度分布如图6所示。图6中,不同颜色表示概率高低,红色区域为高密度分布区,表示分子在该区域出现的概率较大;蓝色区域为低密度分布,表示分子出现概率较低。由图6可知,3种杂原子分子在分子筛中的分布类似,主要分布在超笼中,在方钠石笼(SOD)中也有少许分布。与吡咯和呋喃比较,噻吩在超笼中的分布更加集中(红色区域),同时在SOD笼中的集中区域也更明显。吡咯和呋喃的密度分布比较接近,表明它们在分子筛孔道中的分布类似。这与它们在分子筛中的饱和吸附量和吸附热大小的规律一致。

图6 噻吩、吡咯和呋喃在FAU分子筛中的分布概率密度图Fig.6 Probability density distribution of heteroatom molecules in H-FAU zeolite(a)Thiophene;(b)Pyrrole;(c)Furan
基于上述吸附性质的研究结果我们发现,3种杂原子化合物在分子筛中的饱和吸附量、吸附速率、吸附热大小和孔道中分布密度由大到小的次序均依次为噻吩、吡咯、呋喃,表明在催化裂化条件下3种分子对催化剂分子筛的影响大小依次为噻吩、吡咯、呋喃。
2.4 吸附过程的动力学模拟
由于3种杂原子分子的最小截面尺寸远小于分子筛的孔径,因此杂原子分子在孔道中的扩散性能主要受杂原子类型的影响。在杂原子分子扩散性能的研究中,扩散系数是描述扩散能力、研究传质过程和计算传质速率的重要参数。笔者采用分子动力学方法计算得到了823 K下噻吩、吡咯和呋喃在FAU分子筛中的扩散轨迹,并通过分析扩散轨迹得到了3种分子的均方根位移(MSD)随时间的变化曲线,如图7所示。

图7 噻吩、吡咯和呋喃的MSD曲线Fig.7 MSD curves of heteroatom molecules in H-FAU zeolite
根据爱因斯坦方程(公式(3))计算得到了3种分子在H-FAU分子筛中的扩散系数,见表3。由表3可知,3种杂原子分子在H-FAU分子筛中的自扩散系数大小顺序依次为:呋喃、吡咯、噻吩,即呋喃在分子筛孔道中的扩散阻力最小,噻吩的扩散阻力最大。这与3种分子的吸附模拟结果一致,噻吩的吸附能力强,导致噻吩和孔道的相互作用最大,扩散阻力也最大;而呋喃的吸附能力相对较弱,在孔道中受到束缚相对较小,扩散阻力小于噻吩和吡咯。

表3 噻吩、吡咯和呋喃的扩散系数Table 3 Diffusion coefficients of thiophene,pyrrole and furan
为了进一步考察3种杂原子化合物在H-FAU分子筛上的作用位点,首先计算了3种分子的电荷分布,结果如图8所示。静电势能是指将单位正电荷从无穷远处移到分子周围某处做的功,其中原子核贡献正值,电子贡献负值。从图8中可以看到,3种分子的电荷分布具有一定差异,其中噻吩和呋喃的电荷主要分布在分子环区上、下两侧以及S、O原子附近,并且可以看到,噻吩的电荷分布更加离域,而呋喃的电荷则分布在O原子附近,分布相对局域。这主要是因为O原子的电负性(3.50)比S原子的电负性(2.44)强,导致O原子的吸电子诱导效应更大[5]。吡咯分子电荷主要分布在分子环区两侧。这是因为分子中N—H键偶极矩的方向与p-π共轭方向相同所致。由于化合物分子和分子筛之间的相互作用本质上是电荷的相互作用,因此根据电荷分布可以预测,3种杂原子化合物分子与分子筛相互作用主要通过分子环区和杂原子来实现。

图8 噻吩、吡咯和呋喃分子中的静电势能分布图Fig.8 Electrostatic potential density distribution of heteroatom molecules(a)Thiophene;(b)Pyrrole;(c)Furan;EP—Electronic potential
在扩散计算中,径向分布函数(RDF)表示粒子在空间中的密度变化,是局域密度和平均密度的比,以g(r)表示,r为杂原子或杂原子分子质心与H质子间的距离。笔者采用RDF法,对3种杂原子化合物潜在作用位点(分子环区和杂原子)与H-FAU分子筛孔道结构上的活性位(B酸位)的H质子的间距分布进行统计分析,计算结果如图9所示。
图9中H-Center表示客体分子的质心(Center)在H质子周围的分布;H-X(X=S、O、N)则表示客体分子上的杂原子在H质子周围的分布情况。由3种分子质心的RDF图(图9(a)、(b)、(c))可知,主峰所对应的距离均小于0.4 nm,表明存在分子环区的离域电子与孔道上的H质子相互作用;主峰位置所对应的距离在成键距离范围内说明,杂原子分子和H质子之间可能发生电荷转移。这需要进一步的量化计算。另一方面,由杂原子的RDF图(图9(d)、(e)、(f))可以看到,最强峰的位置同样小于0.4 nm,表明杂原子和H质子之间存在较强相互作用。值得注意的是,呋喃杂原子的RDF峰位置在0.3 nm附近,比噻吩和吡咯的RDF峰位置低,表明分子筛的活性位点(吸附位)对呋喃分子有较强的作用。这主要是因为呋喃分子中的电荷部分局域在电负性较强的O原子附近所导致的,同时这也解释了为什么低压下呋喃的吸附热较噻吩和吡咯的大。综上所述,3种杂原子分子和分子筛上H质子之间存在2种作用形式:(1)分子环区的离域电子和H质子作用;(2)分子上的杂原子和H质子作用。

图9 噻吩、吡咯和呋喃分子与H质子作用的RDF图Fig.9 RDF diagram of thiophene,pyrrole and furan molecules interacting with H protons(a)Thiophene,H-Center distance;(b)Pyrrole,H-Center distance;(c)Furan,H-Center distance;(d)Thiophene,H-S distance;(e)Pyrrole,H-N distance;(f)Furan,H-O distance
3 结 论
在催化裂化反应条件823 K、100~1000 kPa下,3种杂原子化合物分子在分子筛上的饱和吸附量大小、与分子筛的相互作用强弱、对催化裂化反应过程影响由强到弱的顺序都依次为噻吩、吡咯、呋喃;而其在分子筛上的扩散能力强弱顺序依次为呋喃、吡咯、噻吩。
噻吩和吡咯在FAU分子筛上的吸附热随压力的升高而增加,噻吩的热效应比吡咯更强;呋喃表现出与噻吩和吡咯不同变化趋势,其在FAU分子筛上的吸附热随压力的升高而降低,直至吸附平衡时不再变化。
3种杂原子化合物分子在FAU分子筛的超笼和SOD笼结构中均有分布,且噻吩在超笼中的分布较吡咯和呋喃更加集中。3种杂原子化合物分子与分子筛上的H质子相互作用主要通过分子环区的离域电子和杂原子来实现。
