维吉尼亚链霉菌P450-105D1在大肠杆菌中自诱导表达和雄烯二酮的羟化活性
2019-10-16韩毛振罗学才王晶晶周明辉陈亚军
张 雁, 韩毛振, 罗学才, 王晶晶 , 周明辉, 陈亚军
(1. 合肥师范学院 生命科学学院, 合肥 231601; 2. 华中科技大学 生命科学与技术学院, 武汉 430074;3. 合肥停弦渡科技有限公司, 合肥 231601)
近年来,微生物转化在甾体药物的生产中越来越受到重视[1]。雄烯二酮(Androstene)又称AD,是甾体药物生产中重要的中间产物,其甾体母核的羟基化可以大大提高药物活性[2]。雄烯二酮常见的羟基化反应有C9位羟基化[3]和C11位羟基化[4]。其中C9位羟基化产物(9-羟基雄烯二酮)在临床上常用作激素与避孕功能药物使用,同时也是大量甾体药物生产的关键中间产物,其下游产物包括氢化可的松、地塞米松、黄体酮等,具有重要的商业价值。9-羟基雄烯二酮的生产主要使用雄烯二酮为原料进行化学合成,过程繁琐、成本较高,污染严重。而目前发现的分枝杆菌属与红球菌属的部分菌株,其3-甾酮-9α-羟基化酶虽然可以催化雄烯二酮的9位羟基化,但其生物催化过程依然存在菌体有害产物分离、副产品对环境污染、生产效率低而造成生产成本高等问题[5]。
细胞色素P450蛋白(cytochrome P450 proteins/CYP)是一类以亚铁血红素辅因子(heme cofactor)为辅基的超大家族,在各种物种中广泛存在[6]。CYP催化的生物羟化在内源性和外源性分子代谢过程中具有重要的作用[7-10]。CYP105是链霉菌中广泛存在CYP基因家族的重要成员,目前报道的链霉菌全基因组数据中均有发现[11]。CYP105家族基因除了在菌体代谢中的原生功能外,通常存在很宽的底物作用域,表现催化一些对菌体生长无影响的生物转化类型,例如对外源性物质的高效降解[12-13]。维吉尼亚链霉菌(Streptomycesvirginiae)IBL14可以多种甾醇类化合物作为唯一碳源,其次级代谢产生了多种新颖的甾醇类化合物[14-15]。本人在对其催化甾体生物转化研究中发现,基因svu005编码的细胞色素氧化酶CYP105D1可以雄烯二酮为底物,生物催化形成新的化合物。本研究将来自S.virginiaeIBL14的CYP105D1重组蛋白在大肠杆菌中进行自诱导表达,以提高CYP蛋白的活性表达,并对其生物转化雄烯二酮的功能进行验证研究。该催化反应国内外均未见报道,是雄烯二酮微生物转化合成9-羟基雄烯二酮的新途径,可以有效减少合成步骤、降低生产成本。与分枝杆菌属与红球菌属的3-甾酮-9α-羟基化酶催化工艺相比,无菌体有害产物。同时由于CYP蛋白催化的羟化反应具有高度的专一性,可有效减少催化反应副产品,在简化产物纯化工艺、防止生产造成的环境污染方面具有极高的应用潜力。
1 材料和方法
1.1 材料与试剂
S.virginiaeIBL14(CCTCCM 206045)为扩增CYP105D1蛋白编码基因svu005提供模板,质粒pET28a为目的基因的载体,大肠杆菌EscherichiacoliBL21(DE3)为表达菌株,由安徽大学生物集成化实验室提供。
一步法制备大肠杆菌感受态(SSCS)试剂盒(上海捷瑞);DNA凝胶回收试剂盒(上海生工);限制性内切酶NdeI、Hind Ⅲ和T4 DNA连接酶(Takara);质粒小量制备提取试剂盒(上海捷瑞)。
引物合成与基因测序由上海生工完成。高效液相色谱检测所用标准品为:雄烯二酮(Androstene),9-羟基雄烯二酮(9-hydroxy-4-Androstene-3,17-dione),购自上海阿拉丁生化科技有限公司。其他化学试剂均为国产分析纯。
LB培养基(IPTG诱导)以及优化过的ZYM培养基(自诱导)用于重组蛋白的表达研究。通过重组蛋白质的可溶性表达水平比较,选择ZYM培养基作为雄烯二酮生物羟化功能研究的培养基,配方如表1。

表 1 ZYM培养基成分
1.2 svu005的基因分析及分子克隆
以Gene Ontology、KEGG和Swiss-Prot数据库为主要数据库对svu005进行注释,根据注释的结果,进一步从NCBI下载了链霉菌属中已发现的CYP105全部亚家族的代表基因,使用MEGA6.0[16]构建svu005的系统进化树,鉴定其表达蛋白类型。
根据S.virginiaeIBL14全基因组数据,使用Primer Premier5.0进行引物设计。上游引物svu005-F序列为5′-CCCATATGGTGTTGTTCTCCATGTCCGAAGCCC-3′,下划线标注为NdeI酶切位点;下游引物svu005-R序列为5′-CCAAGCTTCACCACGCCACCGGCAGTTC-3′,下划线标注为Hind III酶切位点。
优化后的PCR的扩增程序为95 ℃预变性3 min;94 ℃变性25 s,65 ℃退火25 s,72 ℃延伸1 min,32个循环;72 ℃再延伸10 min。扩增获得的基因片段产物使用1%的琼脂糖凝胶电泳检测,使用DNA凝胶回收试剂盒(上海生工)纯化回收。
1.3 CYP105D1原核表达载体的构建
对svu005的PCR产物和质粒pET28a使用NdeI酶和Hind III酶进行双酶切。酶切产物使用T4连接酶连接,构建表达重组质粒pET28a-svu005。构建过程如图1。重组质粒pET28a-svu005转入采用SSCS试剂盒一步法制备的E.coliBL21 (DE3)感受态细胞,构建工程菌E.coliBL21(DE3)/pET28a-svu005。

图 1 重组质粒pET28a- svu005的构建过程
通过菌落PCR筛选阳性克隆,传代培养6次,使用质粒小量制备提取试剂盒(上海捷瑞)提取质粒,使用NdeI酶进行单酶切验证,同时使用NdeI酶和Hind III酶进行双酶切验证,酶切产物纯化后送生工生物工程(上海)有限公司测序验证。
1.4 CYP105D1蛋白的诱导表达及活性测定
将工程菌E.coliBL21(DE3)/pET28a-svu005和转入空质粒的对照菌株E.coliBL21(DE3)/pET28a分别培养至OD600为0.6时取出,4 ℃保存为种子液。各使用30 mL的含有50 μg/mL的卡那霉素的LB液体培养基和ZYM液体培养基3组,平行接种300 μL种子液,LB液体培养基于30 ℃,220 r/min培养4 h后使用IPTG(终浓度0.5 mmol/L)诱导蛋白表达8 h,ZYM液体培养基于30 ℃,220 r/min条件下培养12 h,并自诱导svu005蛋白表达。重悬于PBS缓冲液中,超声破碎菌体后,10 000 r/min离心10 min,分离可溶性蛋白和沉淀蛋白,对CYP105D1的表达情况进行SDS-PAGE电泳检测。
取离心分离的上清液,使用Lowery法测定上清液总蛋白含量。使用CO差示法测定CYP的表达量,在石英比色皿中加入1.5 mL P450测定液和50 μL连二亚硫酸钠,分别加入20 μL待测上清液,混匀。1 min后使用紫外分光光度仪分别在450 nm和490 nm波长测量基准吸收值。同样的样品通入CO约2 min,稳定1 min分别在450 nm和490 nm波长测量,记录450 nm和490 nm处吸收值,按以下公式计算P450含量。
1.5 CYP105D1蛋白羟化雄烯二酮的功能鉴定
以转入空质粒的菌株E.coliBL21(DE3)/pET28a作为对照,使用ZYM自诱导培养基培养工程菌E.coliBL21(DE3)/pET28a-svu005,在30 ℃,220 r/min条件下培养4 h后,加入1 mL摩尔浓度为1 mmol/L的雄烯二酮(Androstene),反应8 h后,使用乙酸乙酯萃取,加入无水硫酸钠脱水30 min,取上层澄清的乙酸乙酯,37 ℃干燥,1 mL甲醇复溶。使用HPLC(Breeze 1525 series, Waters Co., USA)进行检测,分析柱为250 mm Symmetry C 18(4.6 mm×250 mm, Waters Co., USA),进样体积为10 μL。流动相为乙腈∶水=60∶40,检测波长281 nm。
2 结果和分析
2.1 svu005基因分析及分子克隆结果
PCR扩增获得的svu005序列,经1.0%的琼脂糖凝胶电泳检测,结果见图2。svu005基因全长1224 bp,电泳显示的扩增条带大小相符,目的条带清晰明亮,单一无杂带,说明本次设计的引物可以高效的扩增svu005,扩增结果较为理想。
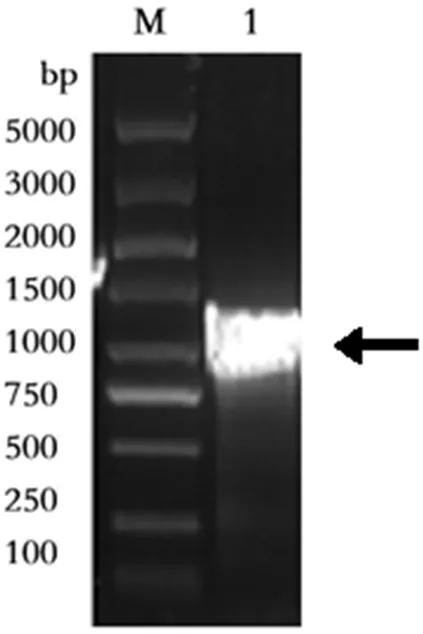
M:DNA ladder;1:svu005基因
图2svu005的扩增结果
Figure 2 PCR products ofsvu005
svu005的表达蛋白Svu005(NCBI:JK119065)被Gene Ontology、KEGG和Swiss-Prot等数据库注释为CYP105D,进一步从NCBI下载了链霉菌属的CYP105亚家族的代表基因。包括Streptomycesgriseolus的CYP105A1(P18326)、CYP105B1(P13827)、CYP105D1(P26911);Streptomycessp.中的CYP105C1(P23296);Streptomyceslavendulae中的CYP105F1(Q9X5P8);Streptomycesnoursei中的Q9L4W8(CYP105H1);Streptomycestenda中的CYP105K1(Q9X9P7);Streptomycesfradiae中的CYP105L1(Q9ZHQ1);Streptomycesclavuligerus中的CYP105M1(Q9KJ93);Streptomycescoelicolor中的CYP105N1(Q9EWP1)和CYP105V1(Q6V1N2);Streptomycesavermitilis中的CYP105P1(Q79ZT4)和CYP105Q1(Q82MP9)和CYP105R1(Q93HF0);Streptomyceshygroscopicus中的CYP105U1(Q84G11);Streptomycesscabies中的CYP105Z1(C9ZGE5);Streptomycestubercidicus的CYP105AA1(Q595R1)。以Streptomycesavermitilis中的CYP1(BAC67818)为外群,使用MEGA6.0[16]基于距离矩阵邻接法构建的svu005表达蛋白的系统进化树,结果如图3。基于以上结果,svu005的表达产物鉴定为CYP105D1。对其表达蛋白的进一步分析发现,svu005编码的CYP105D1含有408个氨基酸,分子量为45.141 62 ku,等电点PI值为5.31,具有CYP蛋白I-helix(G247HETT251)、K-helix(E285LMR288)和Heme binding otif(H346LAFGFGIHQCLG358)3个保守结构域。
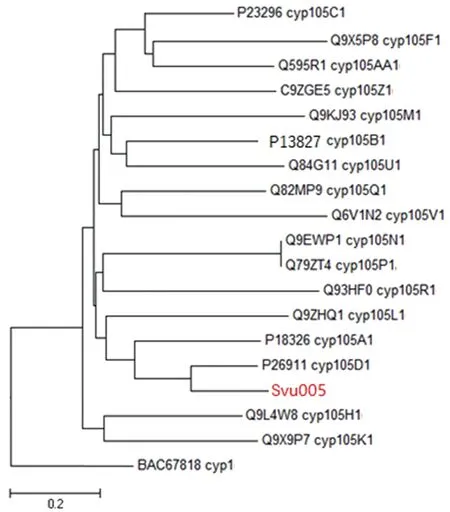
图3 距离矩阵邻接法构建的链霉菌属CYP105家族基因系统进化树
2.2 重组表达质粒pET28a-svu005的构建及验证
将质粒pET28a和svu005扩增序列同时使用NdeI酶和Hind III酶进行双酶切,使用T4连接酶连接,构建重组表达质粒pET28a-svu005。E.coliBL21(DE3)培养至OD600值为0.6时,取1 mL菌液,按照SSCS试剂盒的操作步骤,一步法制得E.coliBL21(DE3)的感受态细胞。将转入重组质粒感受态的E.coliBL21(DE3),取30个菌落进行PCR筛选时,显示出极高的转化效率,阳性克隆占93%。使用质粒小量制备提取试剂盒(上海捷瑞)抽提重组质粒,NdeI酶单酶切验证及NdeI酶/Hind III酶双酶切验证,结果如图4。琼脂糖凝胶电泳结果显示重组质粒pET28a-svu005被NdeI单酶切切开的线性片段长度在6000 bp左右,符合预期大小,重组质粒pET28a-svu005被Hind III和NdeI切成两个长度不等的片段,长度大于4000 bp的大片段为pET28a的双酶切产物,长度在1000 bp至1500 bp之间的小片段为重组基因svu005的双酶切产物,片段大小符合预期。svu005酶切片段回收后送生工生物工程上海有限公司测序,测序结果显示序列正确,无位点突变。
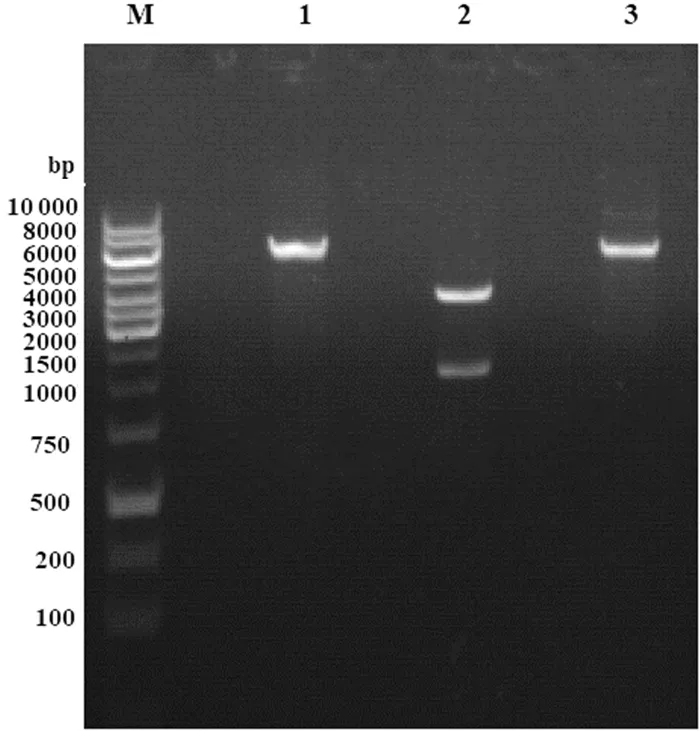
M:3000 bp DNA ladder;1:pET28a-svu005NdeI单酶切结果;2:pET28a-svu005Hind III和NdeI双酶切结果;3:pET28a-svu005原始质粒
图4pET28a-svu005重组质粒酶切验证
Figure 4 Restriction enzyme digestion verification of pET28a-svu005
2.3 CYP105D1重组蛋白的表达及活性
SDS-PAGE检测结果显示LB培养基培养的重组菌株,目的蛋白CYP105D1得到表达,蛋白质分子量与预期的蛋白质分子量45.1 ku大小一致,但是基本上都以不可溶的包涵体形式存在(图5-A)。自诱导ZYM培养基同样存在的包涵体形式表达,但上清液中也有明显的CYP105D1可溶性表达(图5-B)。
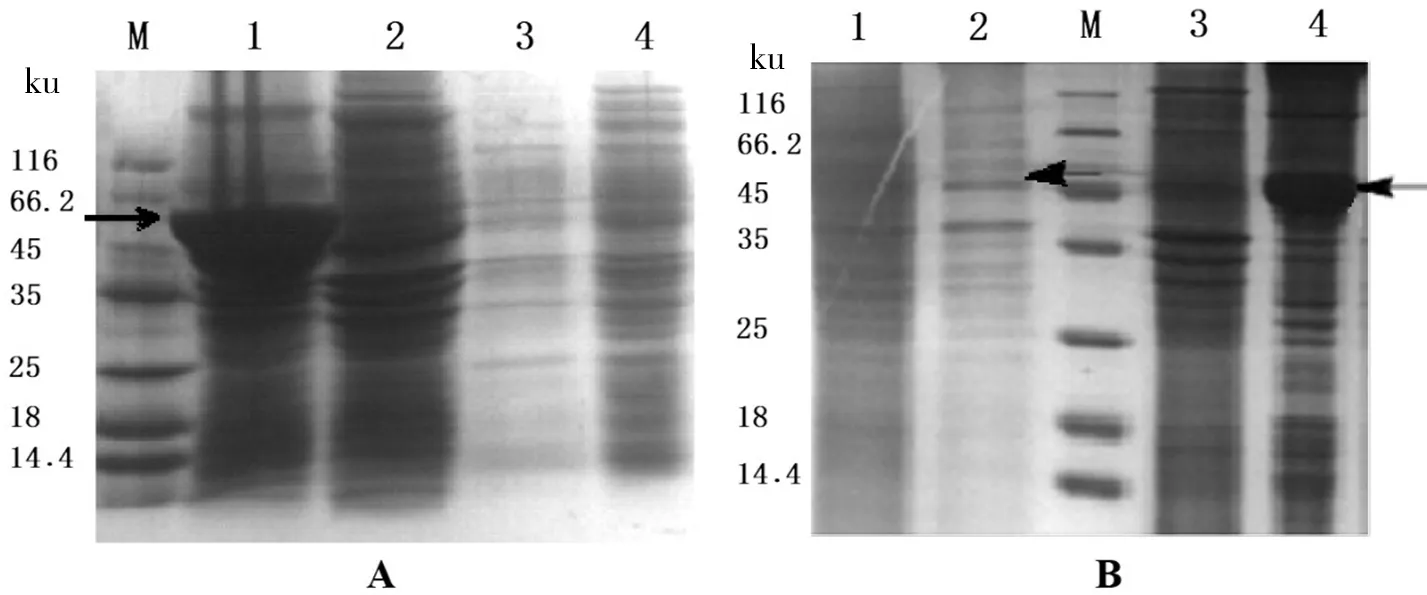
A中,M:蛋白质Marker; 1:E.coliBL21(DE3)/pET28a-svu005诱导破碎后的沉淀; 2:E.coliBL21(DE3)/pET28a诱导破碎后的沉淀; 3:E.coliBL21(DE3)/pET28a-svu005诱导破碎后的上清; 4:E.coliBL21(DE3)/pET28a诱导破碎后的上清。B中,M:蛋白质Marker; 1:E.coliBL21(DE3)/pET28a诱导破碎后的上清; 2:E.coliBL21(DE3)/pET28a-svu005诱导破碎后的上清; 3:E.coliBL21(DE3)/pET28a诱导破碎后的沉淀; 4:E.coliBL21(DE3)/pET28a-svu005诱导破碎后的沉淀
图5(A)LB培养基中IPTG诱导表达重组蛋白CYP105D1的SDS-PAGE检测;(B)ZYM培养基中自诱导表达重组蛋白CYP105D1的SDS-PAGE检测
Figure 5 (A)SDS-PAGE analysis of the CYP105D1 from the recombination strain in LB medium,(B)SDS-PAGE analysis of the CYP105D1 from the recombination strain in ZYM medium
OD600的测量结果表明,ZYM培养基在12 h的培养时间内,菌体产量较高(表2)。活性蛋白的测定结果显示,CYP105D1蛋白在ZYM培养基培养条件下自诱导的CYP蛋白平均产量为0.0916 nmol/L,LB培养基培养条件下使用IPTG诱导的CYP蛋白平均产量为0.0367 nmol/L。自诱导条件下CYP蛋白的可溶性表达量显著性(P= 0.002 208, One way ANOVA检验,Permutaionn= 99 999)高于传统的IPTG诱导方式(表2)。因此,本研究使用ZYM培养基自诱导表达是CYP105D1的优化表达策略,显著提高了CYP105D1蛋白的可溶性表达效率。
2.4 CYP105D1对雄烯二酮的羟化功能鉴定
本研究还尝试过在CYP105D1蛋白的N端添加组氨酸标签,分离纯化CYP105D1蛋白进行功能研究,但是分离的含组氨酸融合标签CYP105D1蛋白不具有生物活性,因此后续功能验证研究均以载入空质粒pET28a的菌株作为对照菌株,使用重组菌株E.coliBL21(DE3)/pET28a-svu005进行菌体内生物催化。使用ZYM培养基自诱导12 h后,svu005表达的CYP105D1蛋白可以有效的羟化雄烯二酮。基于雄烯二酮的C9位是最常见的羟基化位点,使用9-羟基雄烯二酮的标准品检测,其保留时间与E.coliBL21(DE3)/pET28a-svu005以雄烯二酮为底物的羟化产物保留时间一致(图6)。改变不同的洗脱条件,该产物峰的保留时间均与标准品一致,因此初步鉴定羟化产物为9-羟基雄烯二酮。
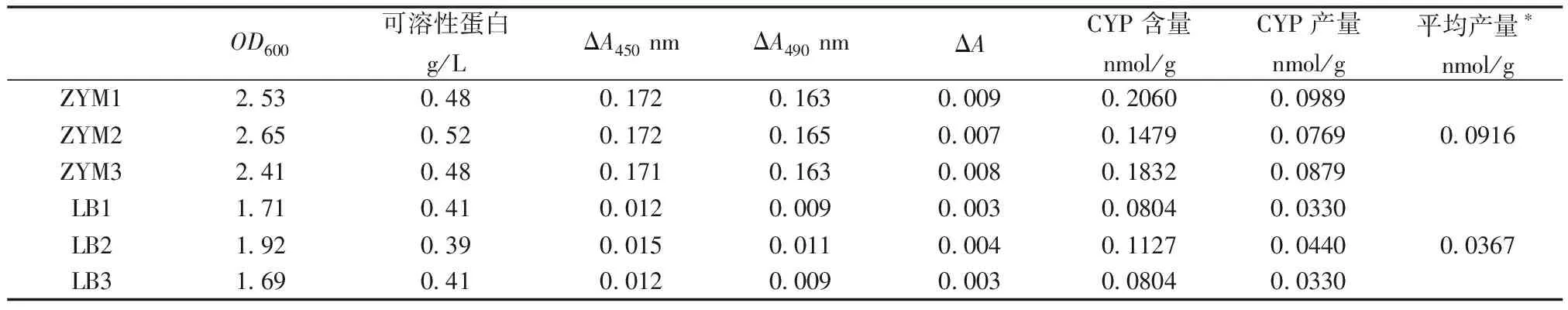
表2 不同诱导方式下CYP105D1产量比较
注:*P值<0.01, One way ANOVA检验,Permutaionn=99 999
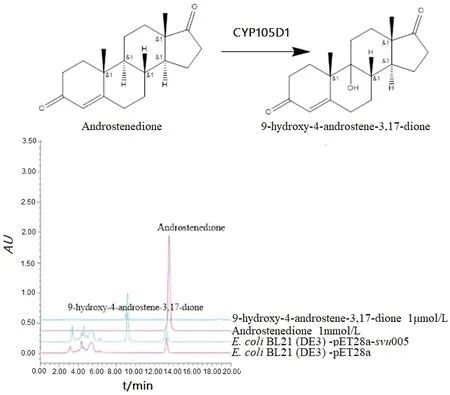
图6 重组菌株E. coli BL21(DE3)/pET28a,E. coli BL21(DE3)/pET28a-svu005生物转化雄烯二酮的HPLC分析
3 讨论
CYP蛋白在重组表达时,其含铁原子的卟啉环辅基[17]是蛋白质正确折叠的关键[18],IPTG的诱导极易形成包涵体,且对菌体具有毒害作用,影响菌体的生长。LB培养基的营养成分较为简单,因此难以实现大肠杆菌的高密度培养[19]。而本研究优化的自诱导培养基营养丰富且不添加IPTG诱导剂,不存在IPTG对细菌的生长的抑制,同时增加可溶性蛋白的表达量[20]。3组平行样品的OD600值显示,使用自诱导培养基,相同培养条件下,重组菌的生长及可溶性蛋白含量均优于使用LB培养基。张彭湃等人使用乳糖代替IPTG诱导P450 BM-3在大肠杆菌中可溶性表达,目标蛋白量提升2.13倍[21],而本研究中,含乳糖成分的自诱导培养基使CYP蛋白的活性表达提升了2.5倍。因此相较于传统的IPTG诱导,本研究采用的自诱导培养是CYP蛋白重组表达的有效培养条件,可以显著提高CYP蛋白的可溶性表达。CYP蛋白的正确折叠需要含铁原子的卟啉环作为辅基,其蛋白质的空间结构折叠较为复杂,将包涵体变性后复性难以重新折叠生成有活性蛋白。另外,CYP催化的羟化反应需要一系列的辅酶还原NADH,以保证反应的持续进行。因此在进一步研究中,可以采用添加卟啉环及其合成前体、添加或共表达P450还原酶辅酶[22-23]等措施进一步提高CYP蛋白的活性表达及转化效率。
CYP基因可以催化各种复杂的反应,包括不同底物的氧化、羟化等,其催化反应具有区域选择性和立体化学型[24]。对于化学方法难以实现羟基化的复杂底物,CYP105家族基因可以在其饱和碳氢键位置引入羟基[25],例如华法林、红霉素、樟脑、苯并芘和7-乙氧基香豆素均可作为被CYP105D1的羟化底物,磺酰脲类可以被CYP105A1和CYP105b1羟化[26-28]。尤其在甾体类药物生产中,CYP105是最重要的羟化酶[29]。其成员CYP105S可以甾醇类物质为底物,合成大环内酯化合物,同时还具有从羟化、氧化到脱烷基化的连续反应[13]。CYP105A1可以连续在维生素D3的两个不同的位点进行特异性羟化[30]。显然CYP105家族蛋白在甾体药物的微生物转化方面具有广泛的应用前景。本研究验证了S.virginiaeIBL14的svu005基因编码的CYP105D1蛋白可以有效地羟化雄烯二酮为9-羟基雄烯二酮,CYP105D1蛋白的催化机理和已发现的分枝杆菌属与红球菌属中的3-甾酮-9α-羟基化酶不同,其羟化产物均一,无明显副产品产生,提供了9-羟基雄烯二酮的微生物转化的新途径。对该生物催化反应的进一步优化,可以改变9-羟基雄烯二酮传统生产的繁琐过程,降低生产成本,改善生产过程对环境的污染,对探索微生物转化在甾体药物的工业生产应用具有重要意义。
